MSA-based pLMs encode evolutionary distance but don’t reliably exploit it –
Published
January 29, 2026
Summary
We characterize how MSA Pairformer encodes phylogenetic relationships. Sequence weights correlate with evolutionary distance, with distinct layers specializing as phylogenetic filters. Yet uniform averaging often outperforms learned weights for contact prediction.
NoteAI usage disclosure
We used Claude (Sonnet 4.5) to help write, clean up, and comment our code; we also used it to review our code and selectively incorporated its feedback. Additionally, we used Gemini (3.0 Pro) to suggest wording ideas and then chose which small phrases or sentence structure ideas to use, write text that we edited, rearrange starting text to fit the structure of one of our pub templates, expand on summary text that we provided and then edited the text it produced, and help copy-edit draft text to match Arcadia’s style.
Purpose
Auditing the evolutionary information encoded by MSA Pairformer is a critical step in understanding the behavior of protein language models (pLMs) that leverage external context in the form of MSAs. In this study, we bridge MSA Pairformer with classical phylogenetics to quantify how the model’s internal sequence weighting maps onto inferred phylogenetic trees. We show that while MSA Pairformer effectively encodes evolutionary relatedness, and does so in specific layers, this signal is a subtle optimization rather than a load-bearing pillar for contact prediction accuracy. This work is intended for biologists and model developers seeking to interpret how evolutionary history is represented within pLMs. It provides a framework for evaluating the “phylogenetic operating range” of MSA-based models and highlights the need for future architectures to selectively gauge input signal reliability.
Introduction
The current trajectory of protein language modeling is dominated by a drive toward massive parameter scaling. Models such as ESM2 (Lin et al., 2023) have achieved strong performance by internalizing the evolutionary statistics of protein families within billions of learned weights. However, as sequence databases expand at a rate that outpaces feasible computational growth, this “memorization” through scaling faces a significant sustainability challenge (Akiyama et al., 2025).
MSA-based architectures, such as MSA Transformer (Rao et al., 2021) and the more recent MSA Pairformer (Akiyama et al., 2025), represent a pivot toward models that utilize external evolutionary context as input. By extracting information directly from multiple sequence alignments (MSAs) passed as input to the model at runtime, these models shift the burden of knowledge from fixed parameters to the specific sequences provided in the MSA, allowing for high performance with significantly fewer model parameters. Yet, this shift introduces a new architectural hurdle: resolving specific structural signals within a divergent alignment without diluting them through phylogenetic averaging1.
1 A limitation in MSA-based models whereby sequences are treated as independent of one another, disregarding their evolutionary relatedness to one another. By treating divergent lineages as if they share an identical structure, this process dilutes subfamily-specific signals, like distinct binding interfaces, which can obscure unique evolutionary constraints of the query sequence.
2 In MSA Pairformer, each MSA has one sequence-of-interest, denoted as the “query.”
MSA Pairformer addresses this via a query-biased attention mechanism, positing that performance is maximized when the model can selectively weight sequences based on their specific evolutionary relevance to the query2. However, evolutionary relevance is vague and lacks a precise metric.
To address this ambiguity, we investigate if evolutionary relatedness (measured as patristic distance3 on a phylogenetic tree) serves as a quantifiable correlate of these sequence weights. While the input MSA implicitly encodes phylogenetic history, it remains to be seen if the model actively leverages this biological signal, or if it instead relies on alternative statistical heuristics to build its representations. Distinguishing between these possibilities is critical for determining if the model’s representations are grounded in evolutionary logic as purported, and if this grounding is a causal driver of downstream performance. While the original paper showed that attention weights could separate subfamilies, this was not a fully-fledged characterization of how sequence weights map onto phylogenetic structure, and was demonstrated for just one protein family. This leaves open the question of how accurately the model’s attention mechanism recapitulates evolutionary relatedness and how broadly these patterns hold across diverse protein families.
3 Patristic distance is the distance between two tips/leaves of a phylogenetic tree, calculated as the sum of branch lengths connecting them. Mathematically, \(d_{ij} = \sum_{b \in P_{ij}} l(b)\), where \(P_{ij}\) is the shortest path through the phylogeny connecting taxa \(i\) and \(j\), and \(l(b)\) is the length of branch \(b\).
In this work, we explore these questions directly. We begin by reanalyzing the response regulator (RR) protein family case study presented by Akiyama et al. (2025), and then expand our investigation to thousands of MSAs of diverse protein families spanning the tree of life. Our goal is to scope out the anatomy of the model’s learning of evolutionary distance and how it relates to downstream performance.
Revisiting the response regulator study
The response regulator (RR) family was the original paper’s showcase for demonstrating the power of query-biased attention. By constructing a mixed MSA of GerE, LytTR, and OmpR subfamilies, the authors showed MSA Pairformer could successfully identify key structural contacts unique to each subfamily.
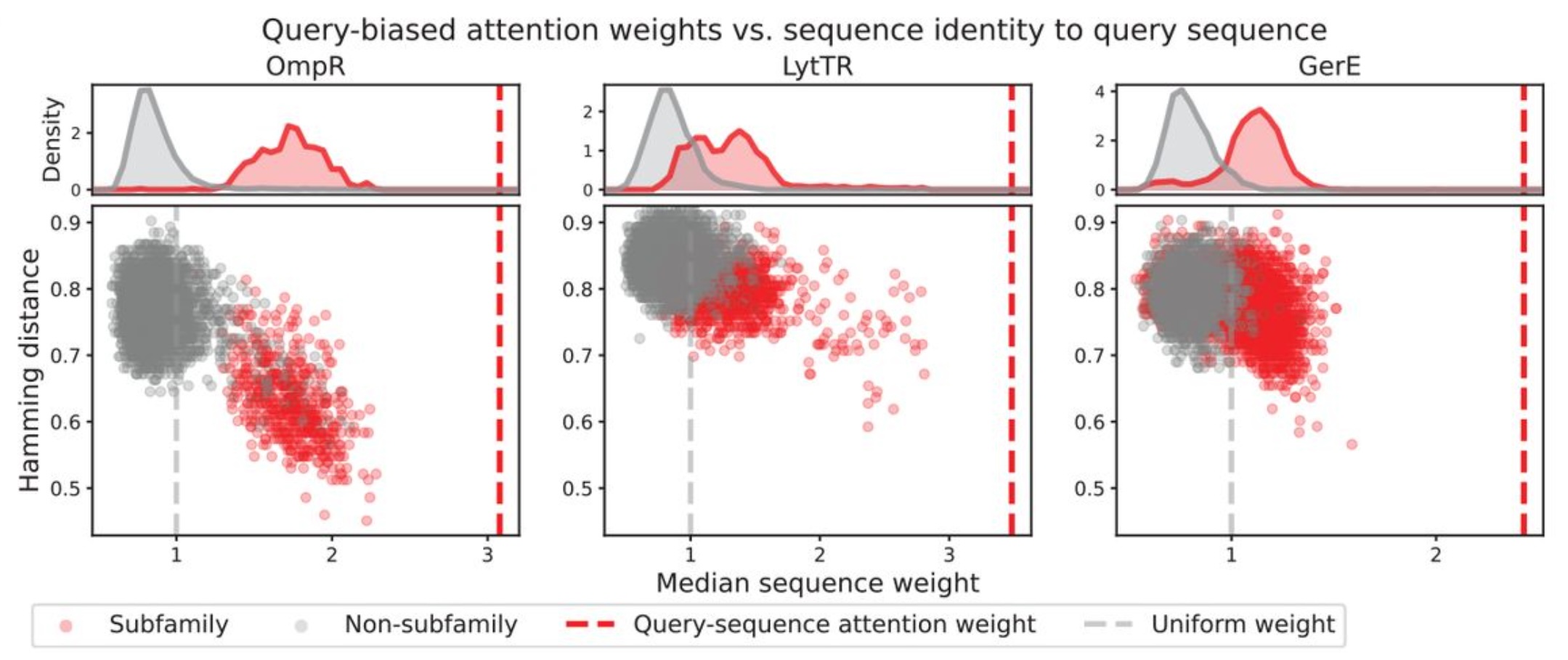
They illustrated one potential mechanism behind this success by plotting model sequence weights against Hamming distance4, revealing that members of the query’s subfamily were consistently upweighted, leading to an inverse correlation between Hamming distance (a proxy for evolutionary distance) and sequence weight, as shown below.
4 Hamming distance is the number of positions at which two sequences of equal length differ.
Figure 4B from Akiyama et al. (2025). Original caption: “Median sequence weight across the layers of the model versus Hamming distance to the query sequence. Top panels show distribution of sequence attention weights for subfamily members (red) and non-subfamily sequences (grey). The grey dotted line indicates weights used for uniform sequence attention and the red dotted line indicates weight assigned to the query sequence.”
We can build on this analysis by replacing the proxy of Hamming distance with a formal phylogenetic tree inferred from the input MSA, which would allow us to ask a more nuanced question: Do the model’s sequence weights recapitulate the continuous evolutionary distances among individual sequences, or do they more closely resemble a binary distinction between “in-group” and “out-group”?
To ground our phylogenetic analysis in the paper’s original findings, we start by replicating the dataset. Following the protocol laid out by Akiyama et al. (2025), we begin by downloading the full PFAM alignments for the GerE, LytTR, and OmpR subfamilies, combining them, and then sampling a final set of 4096 sequences to match the dataset used in the study.
NoteReproducing the response regulator MSAs
Akiyama et al. (2025) qualitatively describe how to reproduce the response regulator MSAs, however these details are insufficient for exact replication. The code below is our attempted reproduction, and we find these MSAs yield similar, yet not identical, sequence weight statistics.
To infer a tree for this MSA, we use FastTree, a rapid method for approximating maximum-likelihood phylogenies suitable for trees of this size (Price et al., 2009).
NoteAccuracy concerns using FastTree
FastTree’s heuristic-based approach to maximum-likelihood inference can sacrifice accuracy compared to more exhaustive methods. We address these concerns in detail in the section, “Is FastTree accurate enough?”, where we benchmark our results against IQ-TREE to quantify the impact of tree inference quality on our findings.
For this MSA, FastTree ends up using the Jones-Taylor-Thornton (JTT) evolutionary model with CAT approximation (20 rate categories). If you’re following along at home, you can check out the full logs at data/response_regulators/PF00072.final.fasttree.log.
Visualizing this tree provides our first direct look at the evolutionary structure of the data.
Tree visualization code
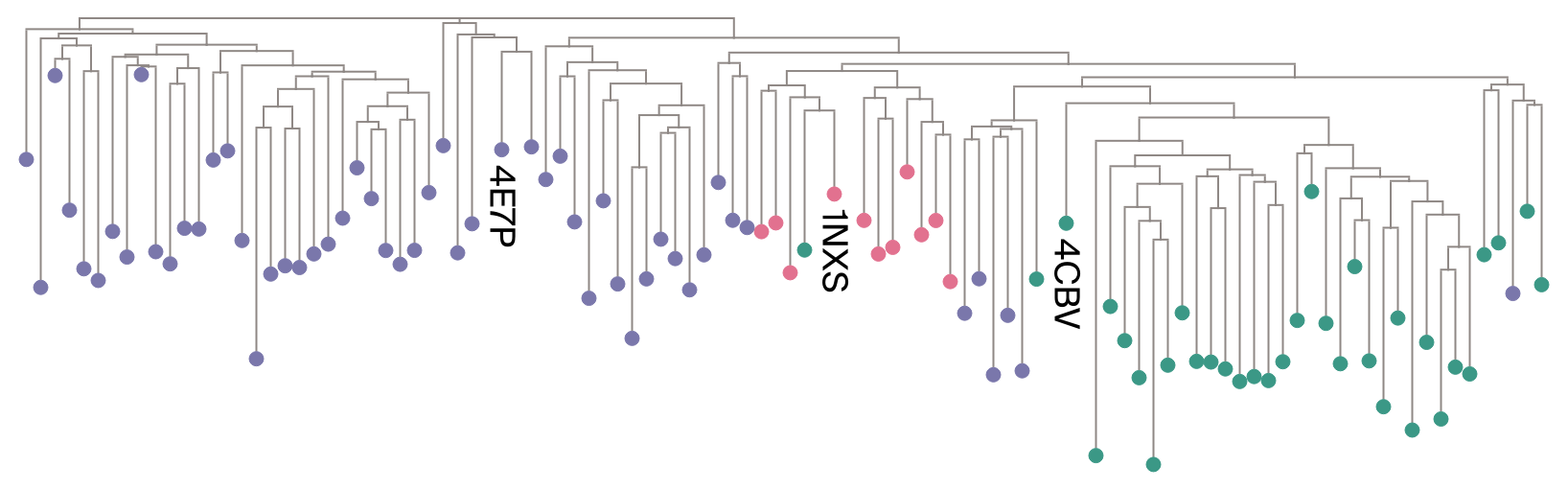
import arcadia_pycolor as apcfrom analysis.plotting import tree_style_with_categorical_annotationfrom analysis.tree import read_newick, subset_treequery_colors = {"4E7P": apc.aster,"1NXS": apc.candy,"4CBV": apc.seaweed,}subfamily_colors = {rr_queries[k]: v for k, v in query_colors.items()}tree_style = tree_style_with_categorical_annotation( categories=target_to_subfamily, highlight=list(rr_queries), color_map=subfamily_colors,)visualized_tree = subset_tree(tree=rr_tree, n=100, force_include=list(rr_queries), seed=42)visualized_tree.render("%%inline", tree_style=tree_style, dpi=300)
Figure 1: A randomly selected subset of the response regulator family illustrating the tree structure. The RCSB structure ID is labelled for the query sequence of each subfamily. Leaves are colored according to subfamily (⬤GerE, ⬤OmpR, ⬤LytTR).
We expect to see largely monophyletic clades corresponding to the three subfamilies, which is more or less what we observe in Figure 1. Discrepancies from this expectation highlight the importance of clade grouping based on explicit tree inference, rather than relying on PFAM domains.
Using the phylogeny as our objective target variable, we assess whether the model’s query-biased sequence weights successfully capture evolutionary relatedness. To do this, we’ll run an MSA Pairformer inference three separate times. Each run will use a different subfamily representative as the query, yielding three distinct, query-biased sets of sequence weights.
NoteRunning MSA Pairformer…
In order to reproduce this step of the workflow, you’ll need a GPU with at least 40GB of GPU VRAM. We don’t assume you have that hardware handy, so we’ve stored pre-computed inference results (data/response_regulators/inference_results.pt), and by default, the code below will load these inference results rather than re-computing. If you have the necessary hardware and want to re-compute the inference, delete this file prior to running the cell below, and the file will be regenerated.
We now have our two key components: a phylogenetic tree for the RR family (our objective target variable) and the model’s sequence weights relative to each of the three subfamily queries. Before diving into a formal statistical analysis, let’s build an intuition for how the model’s attention relates to tree structure by visualizing weights directly onto the tree.
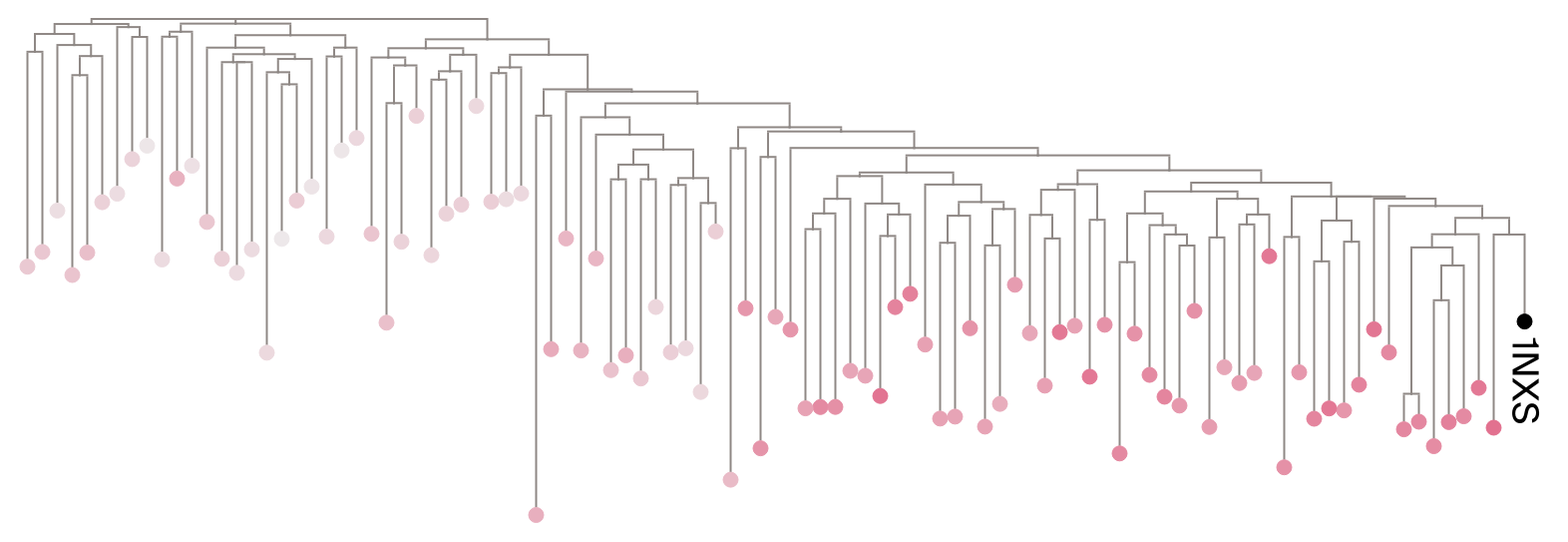
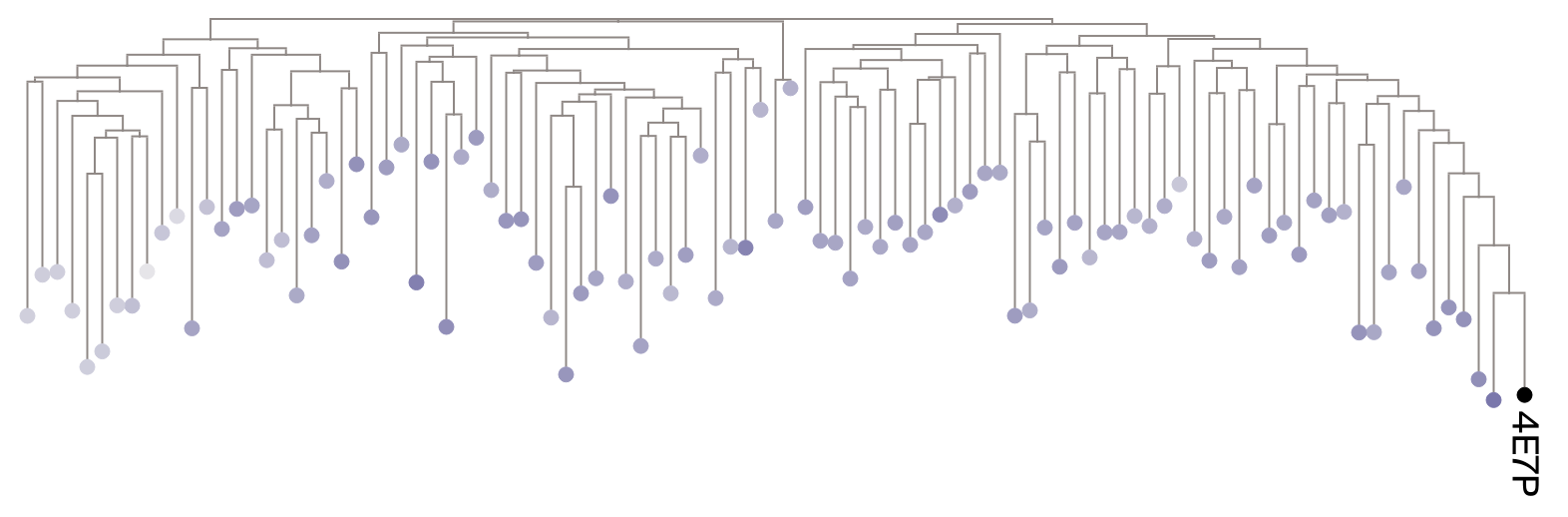
For each of the three queries, let’s center our view on a small subset of the full MSA (for ease of visualization) and color at each leaf the median (across layers) sequence weight it received from the model. If the model is capturing evolutionary relatedness, we’d expect a gradient of sequence weight that follows the tree’s branches away from the query.
(a) Median sequence weights with respect to 1NXS (OmpR).
(b) Median sequence weights with respect to 4CBV (LytTR).
(c) Median sequence weights with respect to 4E7P (GerE).
Figure 2: Unrooted trees for each query, where each leaf is colored according to the median sequence weight it received from the model. Darker nodes signify sequences receiving high levels of attention (upweighting), while lighter nodes signify sequences receiving low levels of attention (downweighting). Each tree is subset to 100 sequences sampled from the full tree (includes all subfamilies). To see whether we observe a gradient, sequences were sampled with a probability inversely proportional to their phylogenetic rank distance from the query, raised to a power of 0.8, which combine to give slight preference for selecting sequences phylogenetically similar to the query while still sampling across the entire phylogeny.
Encouragingly, Figure 2 provides an intuitive picture: weights with respect to 1NXS and 4CBV visually correlate with distance from the query, suggesting the model’s attention mechanism prioritizes evolutionary relatedness. However, the picture is less clear for 4E7P, which invites a quantitative test.
To formalize this observation, let’s extract the patristic distance between the queries and all the other sequences in the MSA, then compare this to the model’s median sequence weights. As in Akiyama et al. (2025), we’ll normalize weights by the number of sequences, so a value of 1 represents the uniform weighting baseline and a value greater than 1 indicates upweighting. And on the suspicion that this median value might smooth over layer-specific complexity, let’s also store the individual weights from each layer to leave room for a more granular, layer-by-layer analysis.
Code
from scipy.stats import linregressfrom analysis.data import get_sequence_weight_datafrom analysis.tree import get_patristic_distancerr_data_dict =dict( query=[], target_subfamily=[], target=[], patristic_distance=[], median_weight=[],)num_layers =22for layer_idx inrange(num_layers): rr_data_dict[f"layer_{layer_idx}_weight"] = []for query in rr_queries: msa = rr_msas[query] targets = msa.ids_l patristic_distances = get_patristic_distance(rr_tree, query) patristic_distances = patristic_distances[targets] weights = get_sequence_weight_data(rr_inference_results[query])# For each layer, sequence weights sum to 1. Scaling by number of# sequences yields a scale where 1 implies uniform weighting. weights *= weights.size(0) median_weights = torch.median(weights, dim=1).valuesfor layer_idx inrange(num_layers): rr_data_dict[f"layer_{layer_idx}_weight"].extend(weights[:, layer_idx].tolist()) rr_data_dict["query"].extend([query] *len(targets)) rr_data_dict["target_subfamily"].extend([target_to_subfamily[target] for target in targets]) rr_data_dict["target"].extend(targets) rr_data_dict["median_weight"].extend(median_weights.tolist()) rr_data_dict["patristic_distance"].extend(patristic_distances.tolist())response_regulator_df = pd.DataFrame(rr_data_dict)response_regulator_df = response_regulator_df.query("query != target").reset_index(drop=True)response_regulator_df
We’ll analyze the relationship between sequence weights and patristic distance with a simple linear regression. We’ll frame the problem to directly assess the explanatory power of the model’s sequence weights: how well do they explain the patristic distance to the query?
For each query \(q\) and each target sequence \(i\) in the MSA, let’s define our model as:
where \(d_{i}\) is the patristic distance from the query \(q\) to the target sequence \(i\), \(w_{i}^{(l)}\) is the normalized sequence weight assigned to sequence \(i\) by a specific layer \(l\), and \(\beta_1^{(l)}\) and \(\beta_0^{(l)}\) are the slope and intercept for the regression at layer \(l\).
Let’s perform this regression independently for each of the three queries. For each query, we’ll calculate the fit using the median weight across all layers and also for each of the 22 layers individually.
We’ll use the coefficient of determination (\(R^2\)) as the key statistic to measure the proportion of the variance in patristic distance that is explainable from the sequence weights. The following code calculates these regression statistics and generates an interactive plot to explore the relationships.
Code
import arcadia_pycolor as apcfrom analysis.plotting import interactive_layer_weight_plotregression_data =dict( query=[], layer=[], r_squared=[], p_value=[], slope=[], intercept=[],)for query in queries_list: query_data = response_regulator_df[response_regulator_df["query"] == query] y = query_data["patristic_distance"].values x = query_data["median_weight"].values result = linregress(x, y) regression_data["query"].append(query) regression_data["layer"].append("median") regression_data["r_squared"].append(result.rvalue**2) regression_data["p_value"].append(result.pvalue) regression_data["slope"].append(result.slope) regression_data["intercept"].append(result.intercept)for layer_idx inrange(num_layers): weight_col =f"layer_{layer_idx}_weight" x = query_data[weight_col].values result = linregress(x, y) regression_data["query"].append(query) regression_data["layer"].append(layer_idx) regression_data["r_squared"].append(result.rvalue**2) regression_data["p_value"].append(result.pvalue) regression_data["slope"].append(result.slope) regression_data["intercept"].append(result.intercept)rr_regression_df = pd.DataFrame(regression_data)rr_regression_dfapc.plotly.setup()interactive_layer_weight_plot(response_regulator_df, rr_regression_df, rr_queries, subfamily_colors)