# pumap_plotting
import scanpy as sc
import anndata as ad
import pandas as pd
import numpy as np
import matplotlib as mpl
import matplotlib.pyplot as plt
import matplotlib.ticker as ticker
from adjustText import adjust_text
import plotly.express as px
import plotly.graph_objects as go
import arcadia_pycolor as apc
import logging
logging.getLogger("scanpy.runtime").setLevel(logging.ERROR)
def setup_plotting_themes():
"""Sets up the plotting themes for matplotlib and plotly."""
apc.mpl.setup()
apc.plotly.setup()
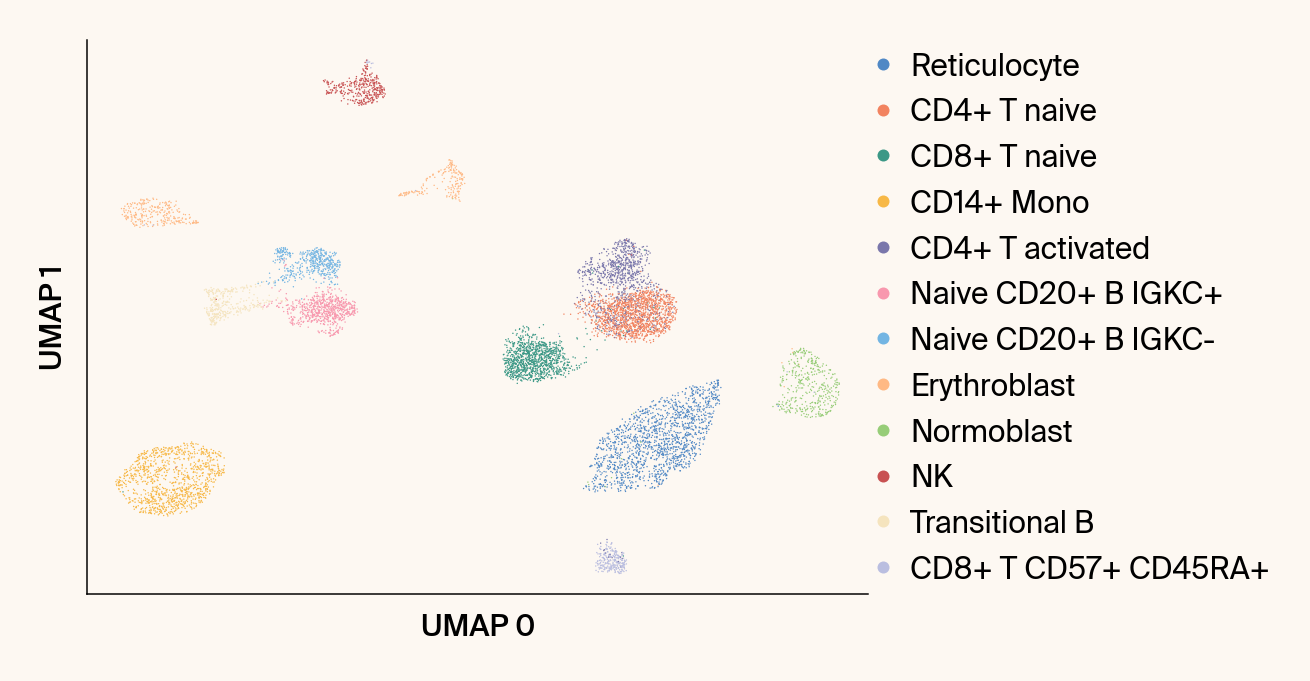
def plot_scanpy_umap(adata: ad.AnnData,
groupby: str = 'cell_type',
n_neighbors: int = 15,
random_state: int = 13):
"""
Computes and plots the standard non-parametric UMAP using Scanpy.
Args:
adata (ad.AnnData): The processed AnnData object.
groupby (str): The .obs column to color by.
n_neighbors (int): Number of neighbors for UMAP.
random_state (int): Random state for UMAP.
"""
if 'neighbors' not in adata.uns:
sc.pp.neighbors(adata, n_neighbors=n_neighbors, use_rep='X_pca')
# Compute standard UMAP
sc.tl.umap(adata, init_pos='random', random_state=random_state)
# Order categories by frequency
category_order = adata.obs[groupby].value_counts().index.tolist()
adata.obs[groupby] = adata.obs[groupby].astype('str').astype(
pd.CategoricalDtype(categories=category_order, ordered=True)
)
with mpl.rc_context({"figure.facecolor": apc.parchment, "axes.facecolor": apc.parchment}):
ax = sc.pl.umap(
adata, color=groupby, size=2,
palette=list(apc.palettes.primary), show=False
)
ax.set_xlabel("UMAP 0")
ax.set_ylabel("UMAP 1")
ax.spines['right'].set_visible(False)
ax.spines['top'].set_visible(False)
ax.set_title("")#f"{groupby} (Standard UMAP)")
plt.show()
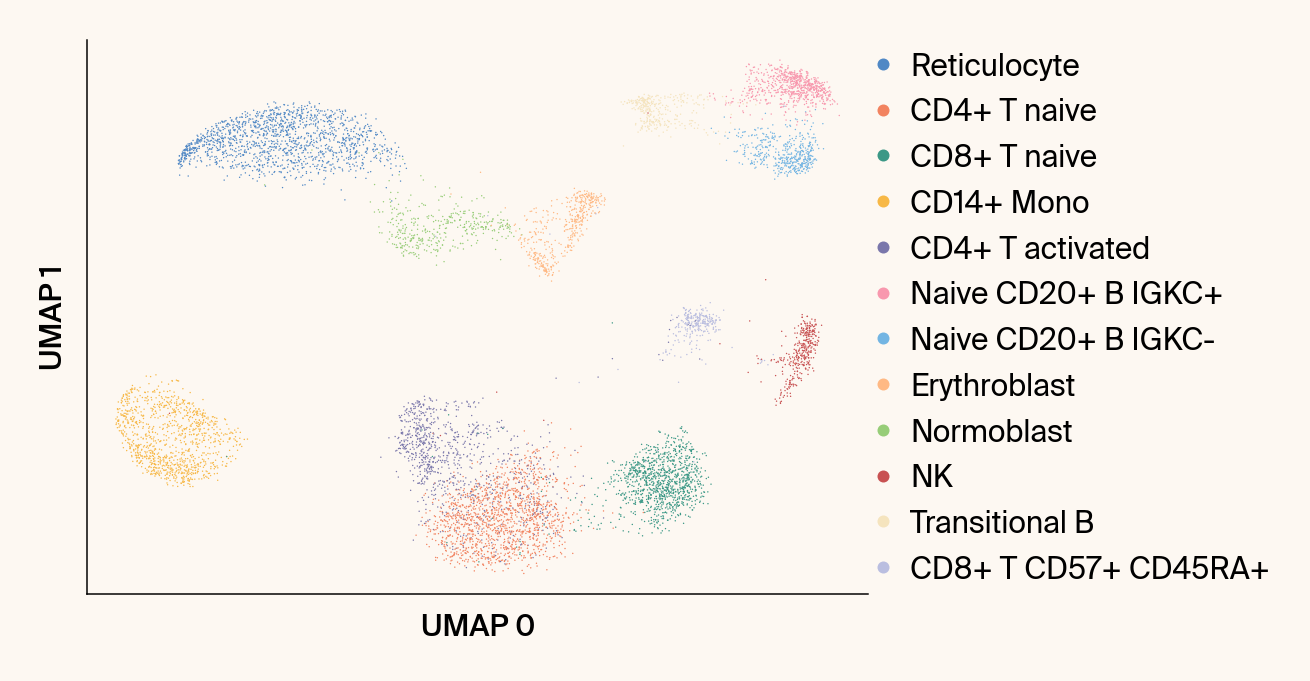
def plot_parametric_umap(reducer: GlassBoxUMAP,
adata: ad.AnnData,
fit_index: int = 0,
groupby: str = 'cell_type'):
"""
Plots the UMAP embedding from a specific parametric model fit.
Args:
reducer (GlassBoxUMAP): The fitted model object.
adata (ad.AnnData): The processed AnnData object.
fit_index (int): The index of the fitted model to visualize.
groupby (str): The .obs column to color by.
"""
if not reducer.embeddings_:
raise RuntimeError("The reducer must be fitted before plotting.")
resolved_index = fit_index if fit_index >= 0 else len(reducer.embeddings_) + fit_index
# Order categories by frequency
category_order = adata.obs[groupby].value_counts().index.tolist()
adata.obs[groupby] = adata.obs[groupby].astype('str').astype(
pd.CategoricalDtype(categories=category_order, ordered=True)
)
# Temporarily assign the parametric embedding to the default UMAP slot
adata.obsm['X_umap'] = reducer.embeddings_[fit_index]
with mpl.rc_context({"figure.facecolor": apc.parchment, "axes.facecolor": apc.parchment}):
ax = sc.pl.umap(
adata, use_raw=False, color=groupby, size=2,
palette=list(apc.palettes.primary),
show=False
)
ax.set_xlabel("UMAP 0")
ax.set_ylabel("UMAP 1")
ax.spines['right'].set_visible(False)
ax.spines['top'].set_visible(False)
ax.set_title("")#
# ax.set_title(f"{groupby} (Parametric UMAP Fit {resolved_index})")
plt.show()
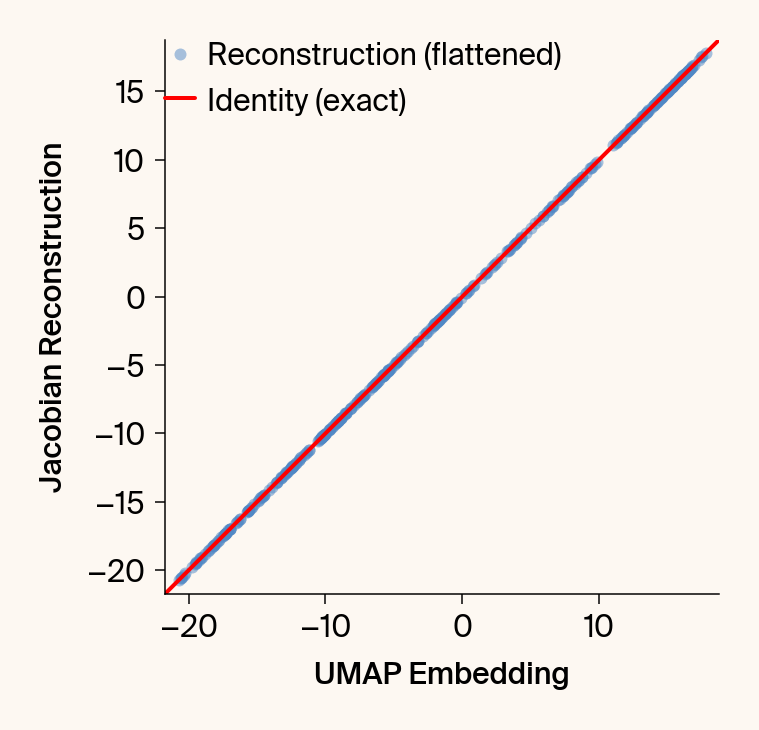
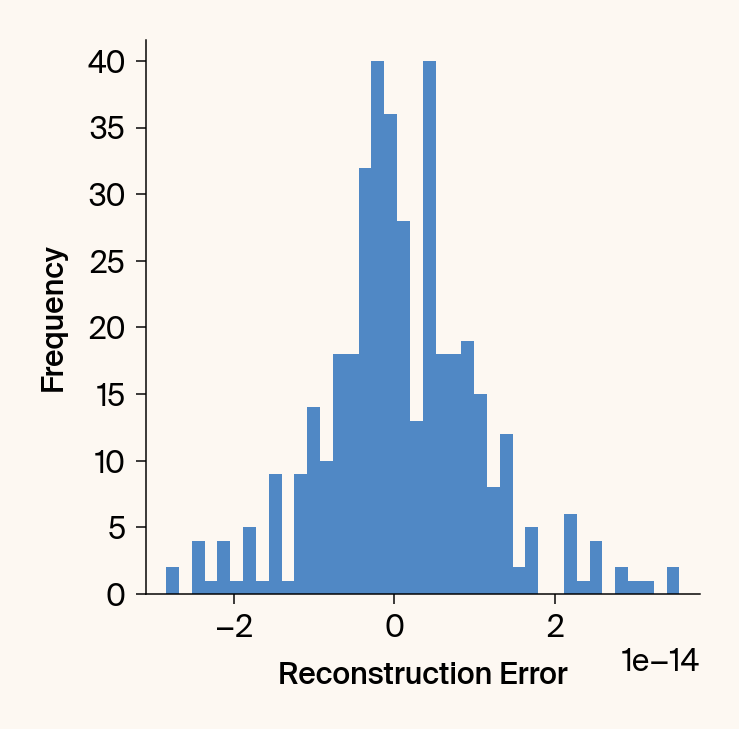
def validate_jacobian(
reducer: 'GlassBoxUMAP', # Use quotes if class is not yet defined
fit_index: int = 0,
n_samples: int = 100,
dtype: torch.dtype = torch.float64
):
"""
Computes and plots the UMAP embedding vs. its reconstruction from the
on-the-fly Jacobian, and plots the reconstruction error.
This function uses the computation method from your 'plot_error' example.
Args:
reducer (GlassBoxUMAP): The fitted model object.
fit_index (int): The index of the fitted model to use.
n_samples (int): The number of samples to use for the validation
(from the start of the training set).
dtype (torch.dtype): The dtype (e.g., torch.float64) for the computation.
"""
if (not reducer.models_ or not reducer.train_data_ is not None):
raise RuntimeError(
"Must run fit() first to have models and training data available."
)
if fit_index >= len(reducer.models_):
raise IndexError("fit_index is out of bounds for reducer.models_.")
device = reducer.device_
if "cuda" not in device:
n_samples = 8
encoder_casted = reducer.models_[fit_index].encoder.to(device=device, dtype=dtype)
encoder_casted.eval()
if n_samples > reducer.train_data_.shape[0]:
n_samples = reducer.train_data_.shape[0]
pca_data = reducer.train_data_[:n_samples]
data_batch_casted = pca_data.to(device=device, dtype=dtype)
jac_batch = torch.autograd.functional.jacobian(
encoder_casted, data_batch_casted, vectorize=True, strategy="reverse-mode"
)
reconstruction = torch.einsum(
'bibj,bj->bi',
jac_batch,
data_batch_casted
)
embedding = encoder_casted(data_batch_casted)
err = reconstruction - embedding
embedding_np = embedding.detach().cpu().numpy()
reconstruction_np = reconstruction.detach().cpu().numpy()
err_np = err.detach().cpu().numpy()
try:
plot_context = mpl.rc_context({
"figure.facecolor": apc.parchment,
"axes.facecolor": apc.parchment
})
except Exception:
print("Warning: 'apc.parchment' not found. Using default plot style.")
plot_context = mpl.rc_context({})
with plot_context:
plt.figure()
plt.scatter(embedding_np[:, :].flatten(), reconstruction_np[:, :].flatten(), alpha=0.5, label="Reconstruction (flattened)")
global_min = min(embedding_np.min(), reconstruction_np.min())
global_max = max(embedding_np.max(), reconstruction_np.max())
# Add a small buffer
min_val = global_min - 1
max_val = global_max + 1
plt.plot([min_val, max_val], [min_val, max_val], 'r', linewidth=2, label="Identity (exact)")
plt.xlabel('UMAP Embedding')#, labelpad=33.8)
plt.ylabel('Jacobian Reconstruction')
plt.legend()
# plt.axis('square')
plt.xlim(min_val, max_val)
plt.ylim(min_val, max_val)
ax1=plt.gca()
# ax1.set_aspect('equal', adjustable='box')
ax1.set_box_aspect(1)
plt.show()
plt.figure()
plt.hist(err_np.flatten(), bins=40)
# current_ax = plt.gca()
# current_ax.xaxis.set_major_locator(plt.MaxNLocator(nbins=5, prune='both'))
plt.xlabel('Reconstruction Error')# (Reconstruction - Embedding)', labelpad=20)
plt.ylabel('Frequency')
ax2=plt.gca()
# ax2.set_aspect('equal', adjustable='box')
ax2.set_box_aspect(1)
# plt.title(f'Histogram of Reconstruction Error ({dtype})')
plt.show()
def plot_interactive(
reducer: GlassBoxUMAP,
adata: ad.AnnData,
groupby: str = 'cell_type',
color_by: str = 'group',
top_n_to_show: int = 16,
show_centroids: bool = False,
fit_index: int = 0,
summary_file: str = "analysis_summary_interactive.csv",
show_percentage: bool = False
):
"""
Generates an interactive Plotly UMAP embedding.
Args:
reducer (GlassBoxUMAP): The fitted model object.
adata (ad.AnnData): The processed AnnData object.
groupby (str): The .obs column to use for grouping (e.g., 'cell_type').
color_by (str): 'group' (to color by `groupby` key) or 'top_gene'.
summary_file (str): Path to the .csv file for loading/saving.
"""
import os
if summary_file and os.path.exists(summary_file):
df = pd.read_csv(summary_file)
if groupby not in df.columns:
print(f"Warning: Column '{groupby}' not found in {summary_file}.")
potential_cols = [col for col in df.columns if
col not in ['UMAP 0', 'UMAP 1'] and
not col.startswith('gene_')]
if len(potential_cols) == 1:
old_groupby = potential_cols[0]
df = df.rename(columns={old_groupby: groupby})
else:
raise KeyError(
f"Column '{groupby}' not found in {summary_file}. "
f"Found potential group columns: {potential_cols}. "
"The summary file may be stale. "
"Try running with TRAIN=True to regenerate it."
)
else:
if not reducer.feature_contributions_:
raise RuntimeError("Must run compute_attributions() first to generate data.")
df = reducer._prepare_plotly_df(adata, groupby, fit_index=fit_index)
if summary_file:
df.to_csv(summary_file, index=False)
hover_data = ['gene_0', 'gene_1', 'gene_2', groupby]
if color_by == 'group':
fig = px.scatter(
df, x='UMAP 0', y='UMAP 1', color=groupby,
# title=f'Bone Marrow Gene Expression: {groupby}',
hover_data={k: True for k in hover_data if k != groupby},
category_orders={groupby: df[groupby].astype('category').value_counts().index},
color_discrete_sequence=(apc.palettes.primary + apc.palettes.secondary)
)
grouping_col, data_for_centroids = groupby, df
elif color_by == 'top_gene':
df['gene_0'] = df['gene_0'].astype(str)
top_genes = df['gene_0'].value_counts().nlargest(top_n_to_show).index
df_filtered = df[df['gene_0'].isin(top_genes)]
percent_shown = len(df_filtered) / len(df)
# title = f'Bone Marrow: Top Gene Contributors, ({percent_shown:.1%} of cells shown)'
fig = px.scatter(
df_filtered, x='UMAP 0', y='UMAP 1', color='gene_0',
title="", hover_data=hover_data,
category_orders={'gene_0': top_genes},
color_discrete_sequence=(apc.palettes.secondary + apc.palettes.primary)
)
grouping_col, data_for_centroids = 'gene_0', df_filtered
else:
raise ValueError("color_by must be 'group' or 'top_gene'")
if show_centroids:
centroids = data_for_centroids.groupby(grouping_col)[['UMAP 0', 'UMAP 1']].mean()
for label, center in centroids.iterrows():
fig.add_annotation(
x=center['UMAP 0'], y=center['UMAP 1'], text=f"<b>{label}</b>",
showarrow=False, font=dict(size=16, color='black'),
align='center', bgcolor='rgba(255, 255, 255, 0.5)', borderpad=4
)
fig.update_traces(marker_size=3)
fig.update_layout(
autosize=True,
yaxis_scaleanchor="x",
legend={'itemsizing': 'constant', 'y': 1, 'x': 1.0, 'yanchor': 'top', 'xanchor': 'left'}
)
apc.plotly.style_plot(fig, monospaced_axes="all")
fig.show()
if color_by == 'top_gene' and show_percentage:
print(f'Bone Marrow: Top Gene Contributors, ({percent_shown:.1%} of cells shown)')
def plot_feature_importance_by_group(
reducer: GlassBoxUMAP,
adata: ad.AnnData,
groupby: str = 'cell_type',
n_features_bars: int = 12,
n_features_vectors: int = 3,
fit_index: int = 0,
groups_to_plot: list = None,
summary_stats_file: str = "analysis_summary_stats.csv",
summary_plot_file: str = "analysis_summary_plot_data.npz",
set_axes_equal: bool = False,
plot_sum_features: bool = False
):
"""
Analyzes and visualizes feature contributions for each group.
Args:
reducer (GlassBoxUMAP): The fitted model object.
adata (ad.AnnData): The processed AnnData object.
groupby (str): The .obs column to use for grouping (e.g., 'cell_type').
groups_to_plot (list): A list of specific group names to plot.
If None, plots the top 12.
"""
import os
# Add necessary imports that were implicit in the original
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import matplotlib as mpl
from adjustText import adjust_text
# Assuming 'apc' is available in the environment (e.g., import anndata_plotting_context as apc)
# Assuming 'ad' (anndata) is available
can_load_from_file = (
summary_stats_file and os.path.exists(summary_stats_file) and
summary_plot_file and os.path.exists(summary_plot_file)
)
if can_load_from_file:
summary_df = pd.read_csv(summary_stats_file)
with np.load(summary_plot_file, allow_pickle=True) as data:
embedding = data['embedding']
try:
group_labels_array = data['group_labels']
loaded_groupby_key = str(data['group_by_key']) if 'group_by_key' in data else 'cell_type'
except KeyError:
print("Warning: 'group_labels' key not found. Trying fallback 'cell_types'...")
try:
group_labels_array = data['cell_types']
loaded_groupby_key = 'cell_type'
except KeyError:
raise KeyError(
"Could not find 'group_labels' or 'cell_types' in the .npz file. "
"The summary file may be stale or corrupt. "
"Try running with TRAIN=True to regenerate it."
)
mean_jacobian_vectors = data['mean_jacobian_vectors'].item()
gene_names_original_order = data['gene_names']
if loaded_groupby_key != groupby:
print(f"Warning: File was grouped by '{loaded_groupby_key}', "
f"but you requested '{groupby}'. Results may be incorrect.")
all_groups = pd.Series(group_labels_array).value_counts().index
else:
if not reducer.feature_contributions_:
raise RuntimeError("Must run compute_attributions() first to generate data.")
embedding = reducer.embeddings_[fit_index]
group_labels_array = adata.obs[groupby].values
all_groups = adata.obs[groupby].value_counts().index
gene_names_original_order = adata.var_names.values
summary_df = reducer.get_feature_importance(adata, groupby, gene_names_original_order)
mean_jacobian_vectors = {}
for group in all_groups:
is_group_mask = (adata.obs[groupby] == group).values
mean_jacobian_vectors[group] = np.mean(
reducer.feature_contributions_[fit_index][is_group_mask], axis=0
)
if groups_to_plot is None:
groups_to_plot = all_groups[:12]
# This assumes 'apc' is an imported module available in the scope
cmap = (apc.palettes.primary + apc.palettes.secondary).to_mpl_cmap()
category_colors = [cmap(i / len(all_groups)) for i in range(len(all_groups))]
color_map = {name: color for name, color in zip(all_groups, category_colors)}
point_colors = np.array([color_map.get(ct, 'gray') for ct in group_labels_array])
gene_to_original_index = {gene: i for i, gene in enumerate(gene_names_original_order)}
with mpl.rc_context({"figure.facecolor": apc.parchment, "axes.facecolor": apc.parchment}):
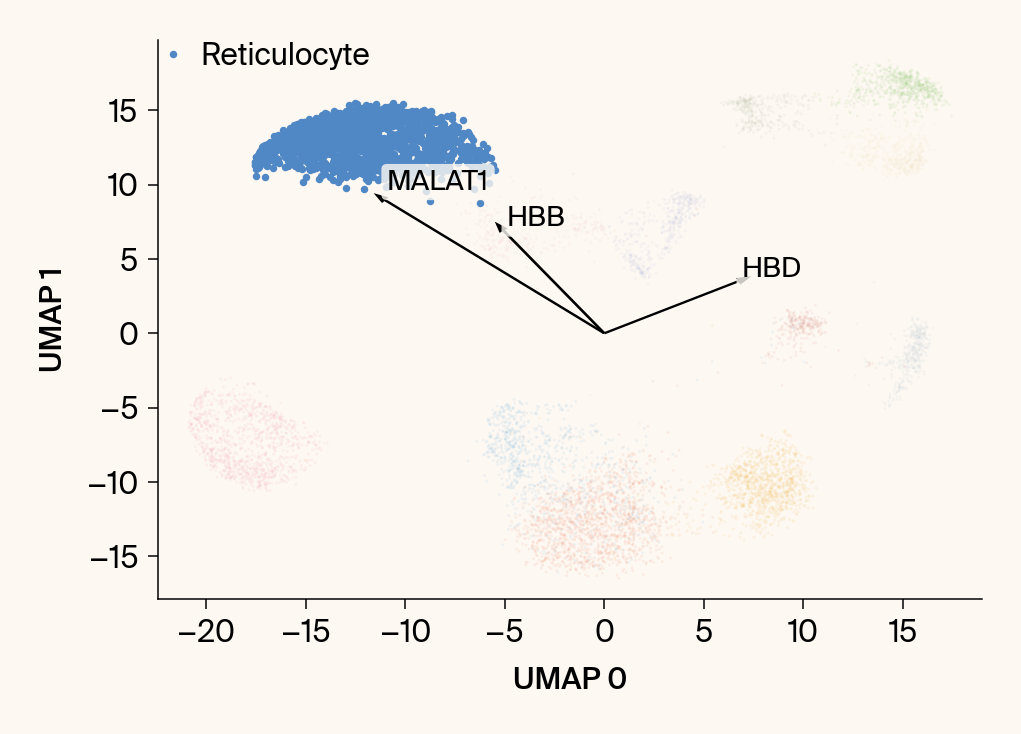
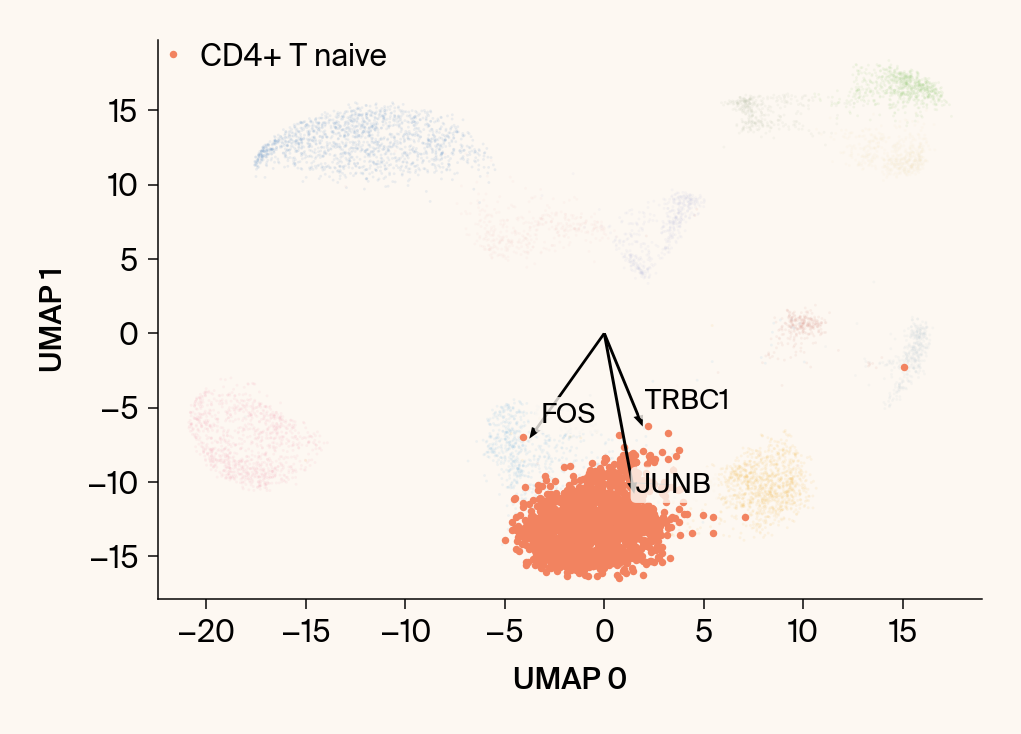
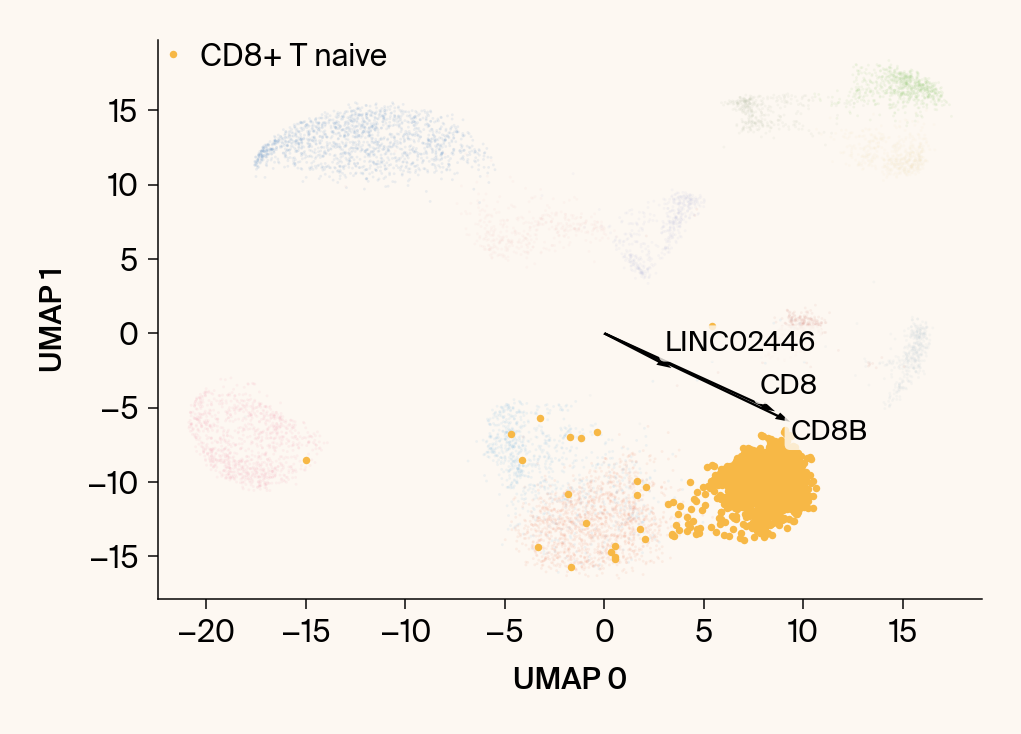
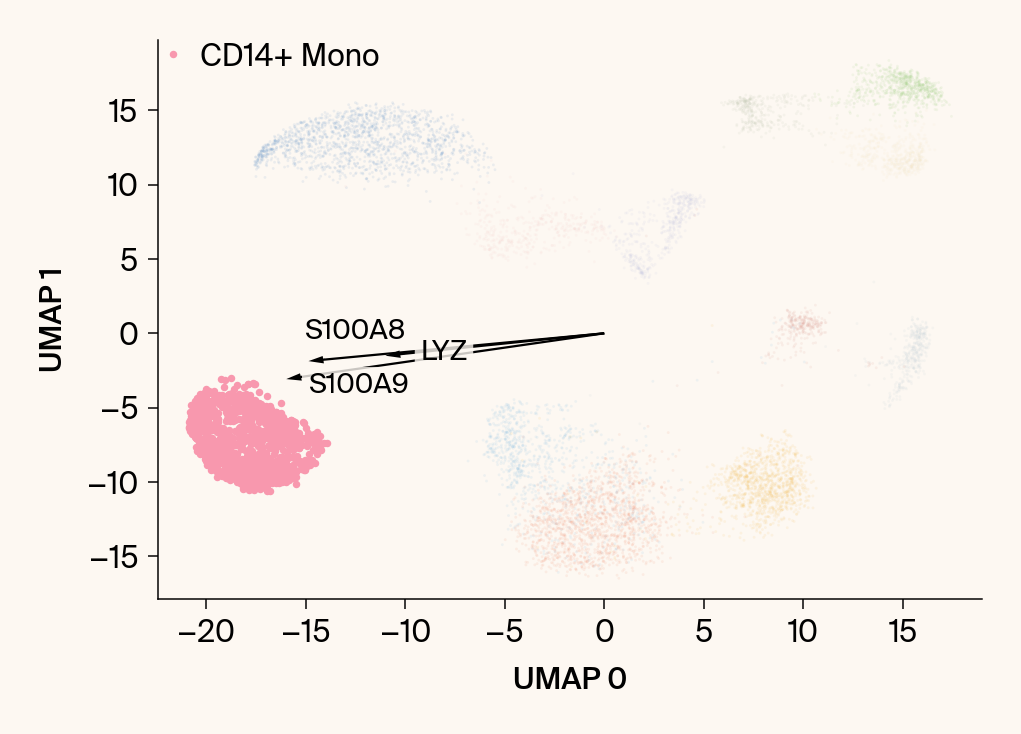
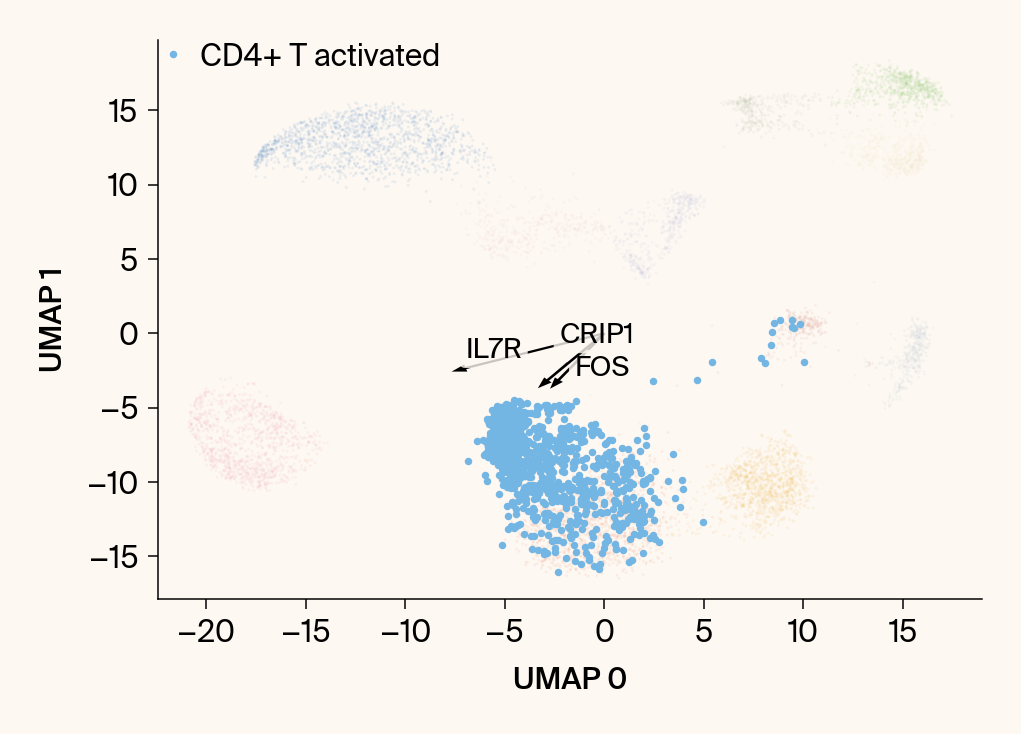
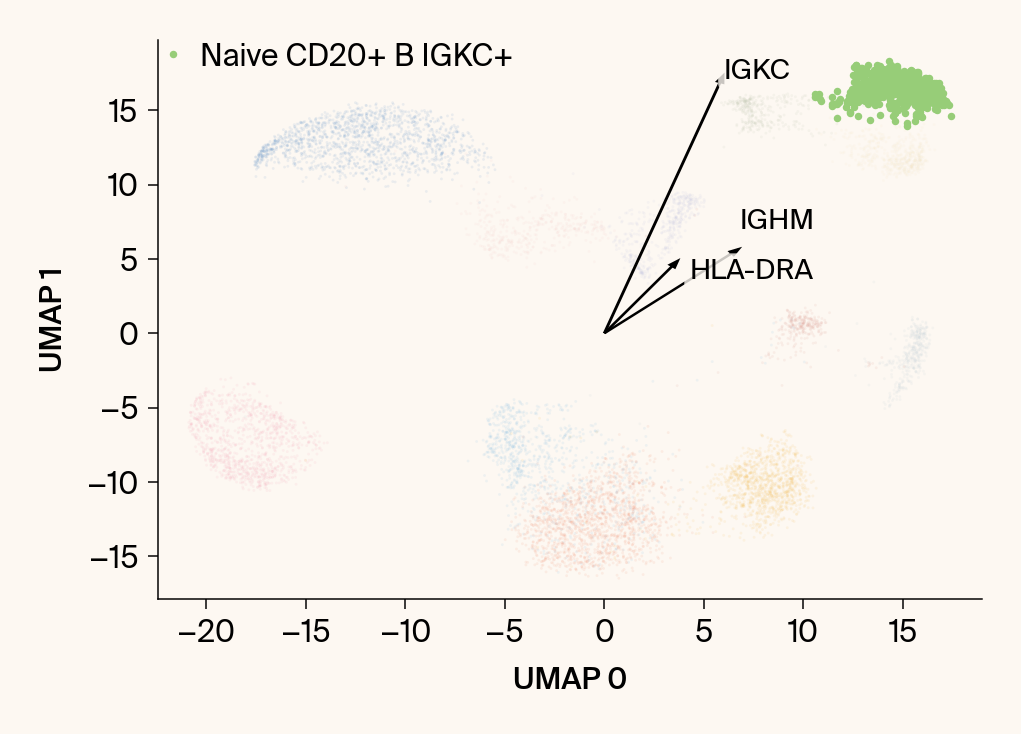
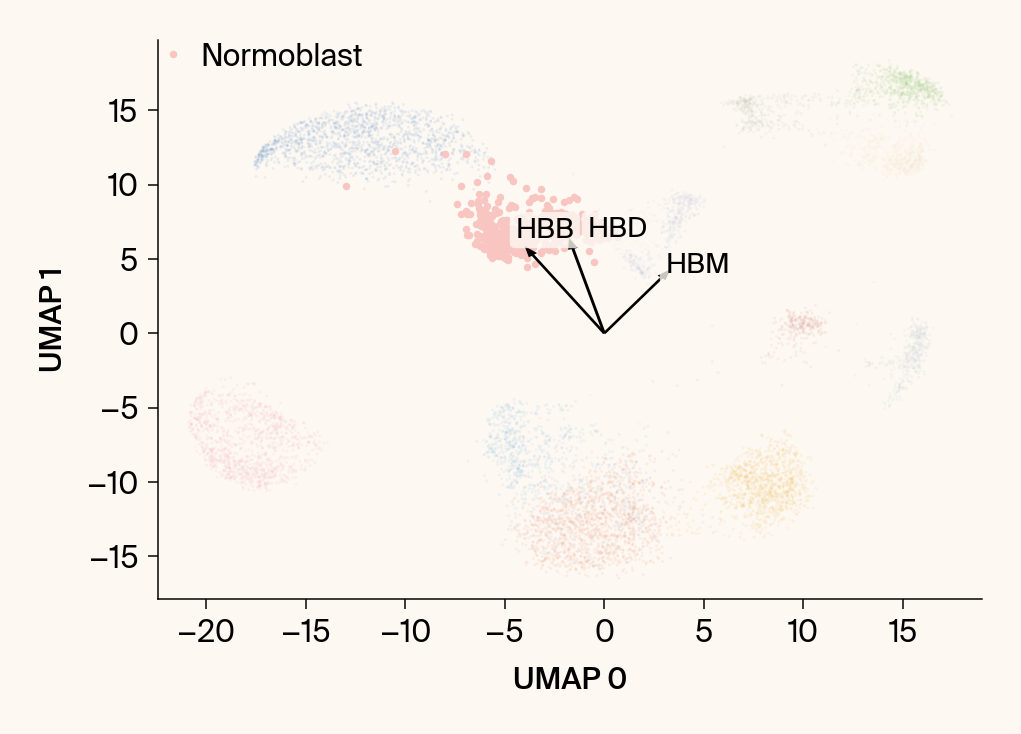
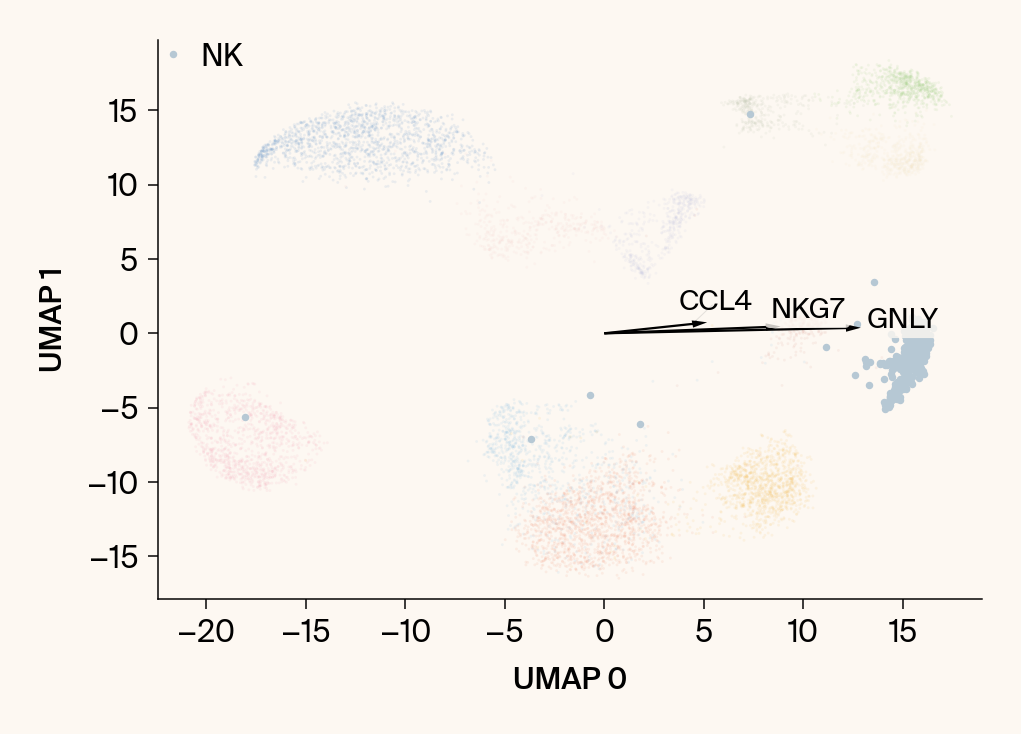
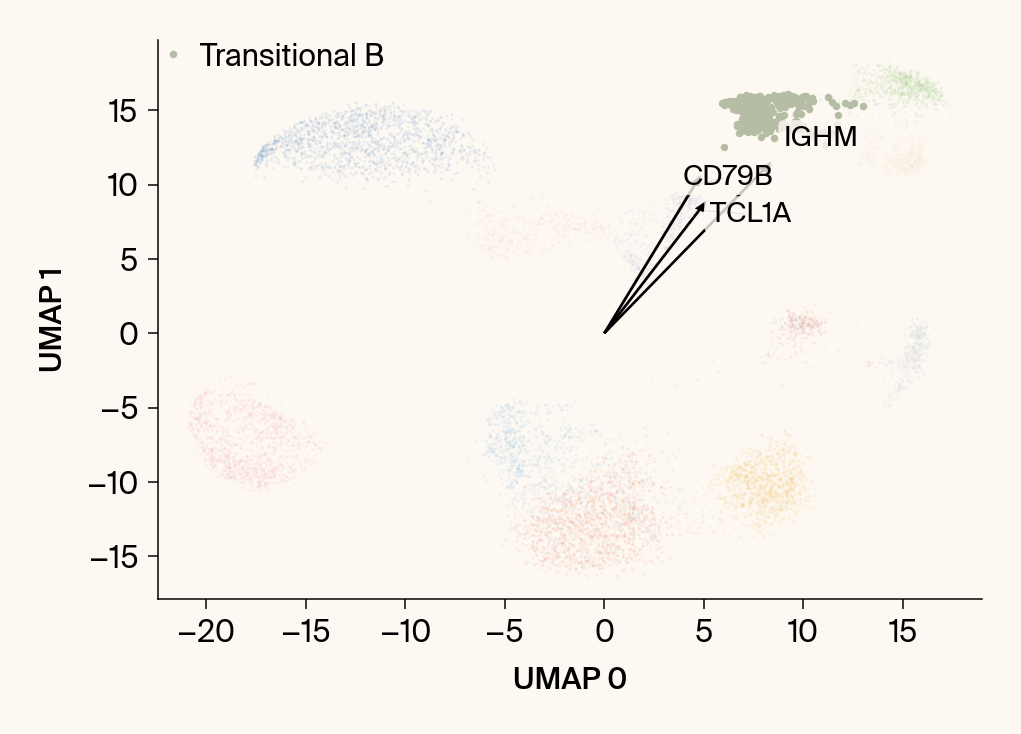
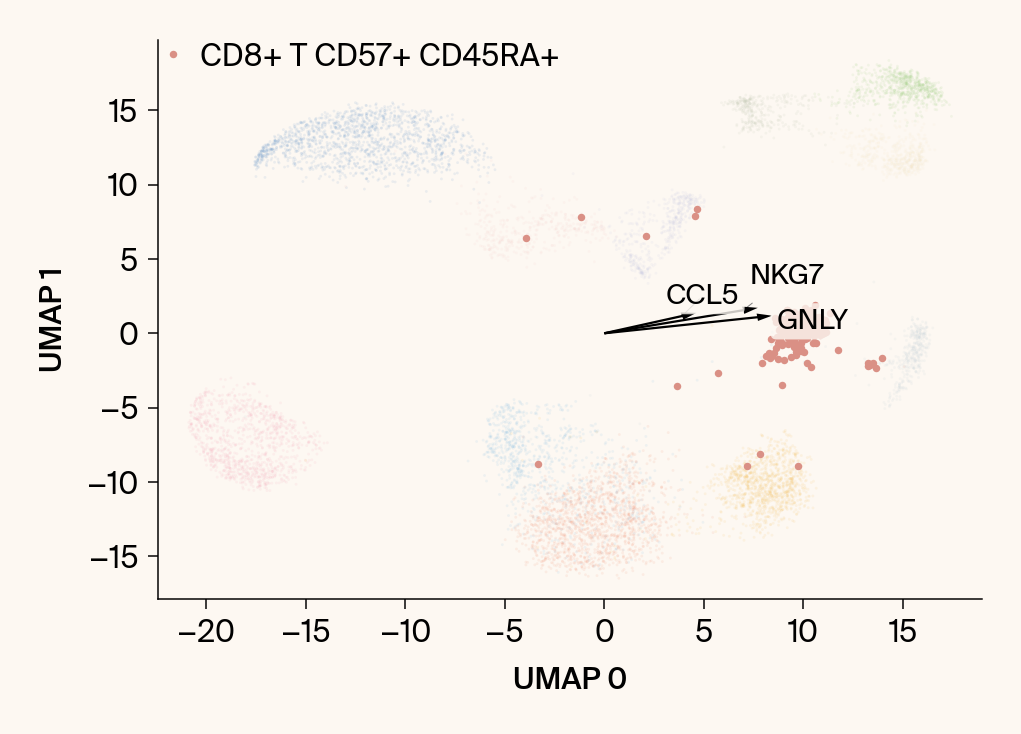
for group in groups_to_plot:
# --- Plot 1: Scatter plot with vectors ---
# Create new figure without subplots
fig1 = plt.figure()#figsize=[6, 6])
is_group_mask = (group_labels_array == group)
# Use plt. scatter, xlabel, ylabel, grid, legend
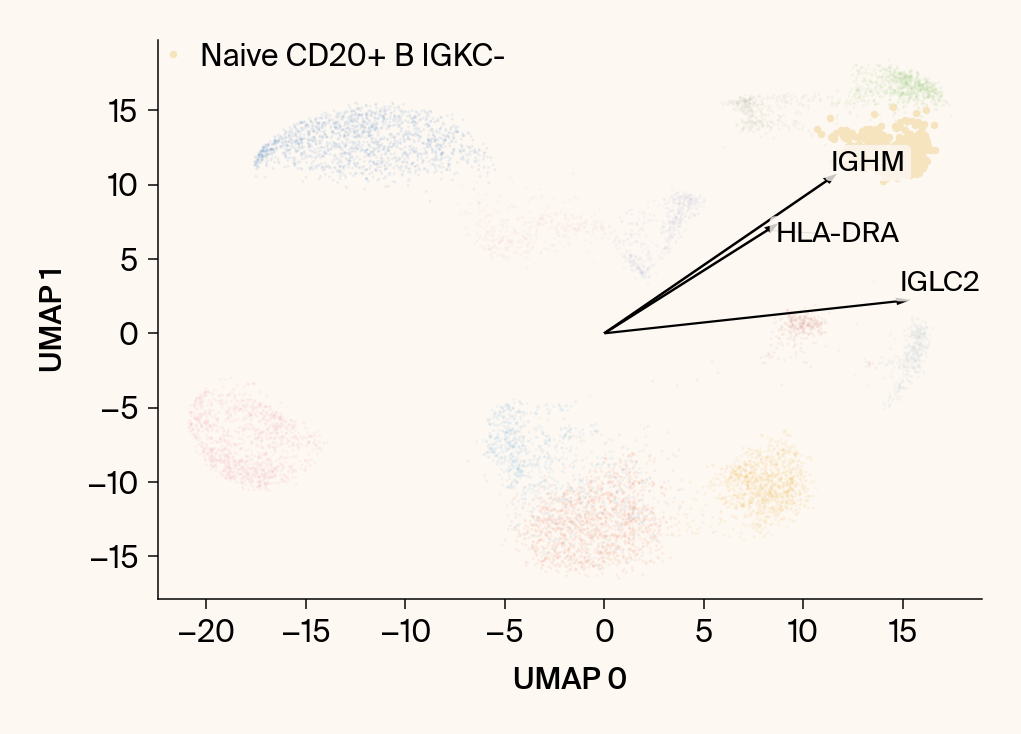
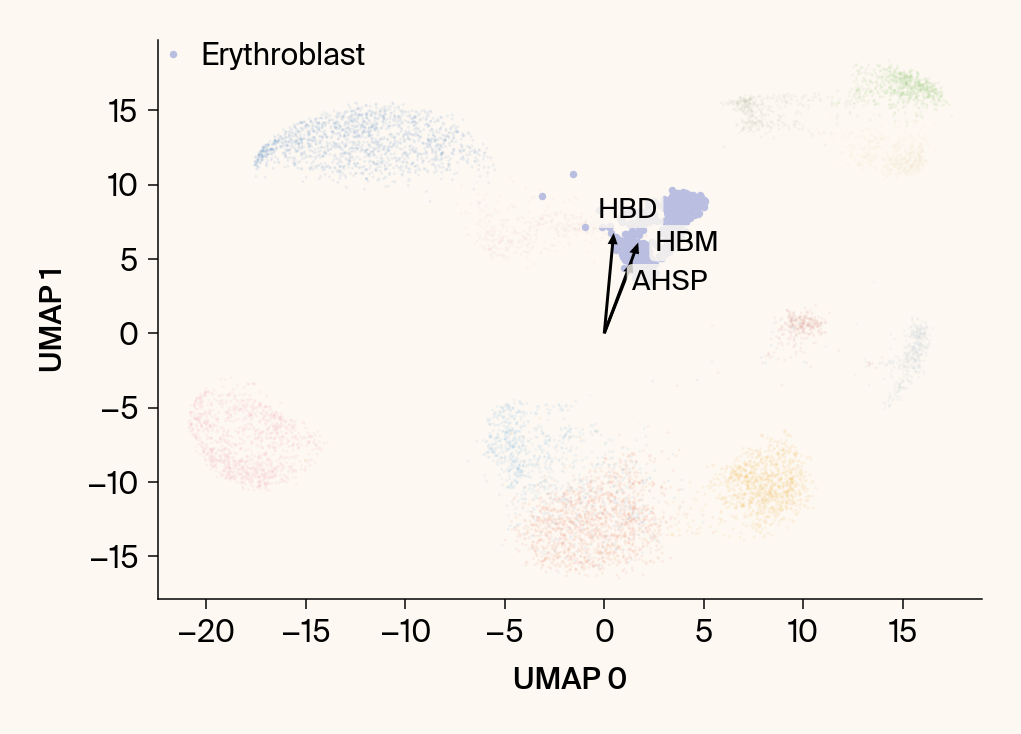
plt.scatter(embedding[:, 0], embedding[:, 1], c=point_colors, s=2, alpha=0.1)
plt.scatter(embedding[is_group_mask, 0], embedding[is_group_mask, 1],
c=[color_map.get(group, 'gray')], s=14, marker="o", label=group)
plt.xlabel("UMAP 0"); plt.ylabel("UMAP 1")
plt.grid(False); plt.legend()
# Get current axis for commands that require an axis object
current_ax = plt.gca()
current_ax.spines[['right', 'top']].set_visible(False)
if set_axes_equal:
current_ax.set_box_aspect(1)
group_df = summary_df[summary_df[groupby] == group].sort_values('mean_contribution', ascending=False)
if group_df.empty:
print(f"Skipping plot for {group}: no data found in summary.")
plt.close(fig1) # Close the empty figure
continue
top_bar_indices = group_df.index[:n_features_bars]
top_vector_indices = group_df.index[:n_features_vectors]
vectors_for_group = mean_jacobian_vectors.get(group)
if vectors_for_group is None:
print(f"Skipping vectors for {group}: no mean_jacobian_vectors found.")
continue
cluster_centroid = np.mean(embedding[is_group_mask], axis=0)
top_gene_names = group_df.loc[top_vector_indices, 'gene']
top_gene_original_indices = [gene_to_original_index[gene] for gene in top_gene_names if gene in gene_to_original_index]
if top_gene_original_indices:
max_vector_mag = np.max(np.linalg.norm(vectors_for_group[:, top_gene_original_indices], axis=0))
scale_factor = (np.linalg.norm(cluster_centroid) / (max_vector_mag + 1e-6)) * 0.8
else:
scale_factor = 1.0
texts = []
for gene_idx in top_vector_indices:
gene_name = group_df.loc[gene_idx, 'gene']
gene_original_index = gene_to_original_index.get(gene_name)
if gene_original_index is not None:
vec = vectors_for_group[:, gene_original_index] * scale_factor
# Use plt.arrow and plt.text
plt.arrow(0, 0, vec[0], vec[1], width=0.15, color='k', head_width=0.5, zorder=3)
texts.append(plt.text(vec[0], vec[1], gene_name, fontsize=14,
bbox=dict(boxstyle="round,pad=0.2", fc=apc.parchment, ec="none", alpha=0.8)))
if texts:
# adjust_text requires an explicit axis
adjust_text(texts, ax=current_ax, arrowprops=dict(arrowstyle="-", color='gray', lw=0.5))
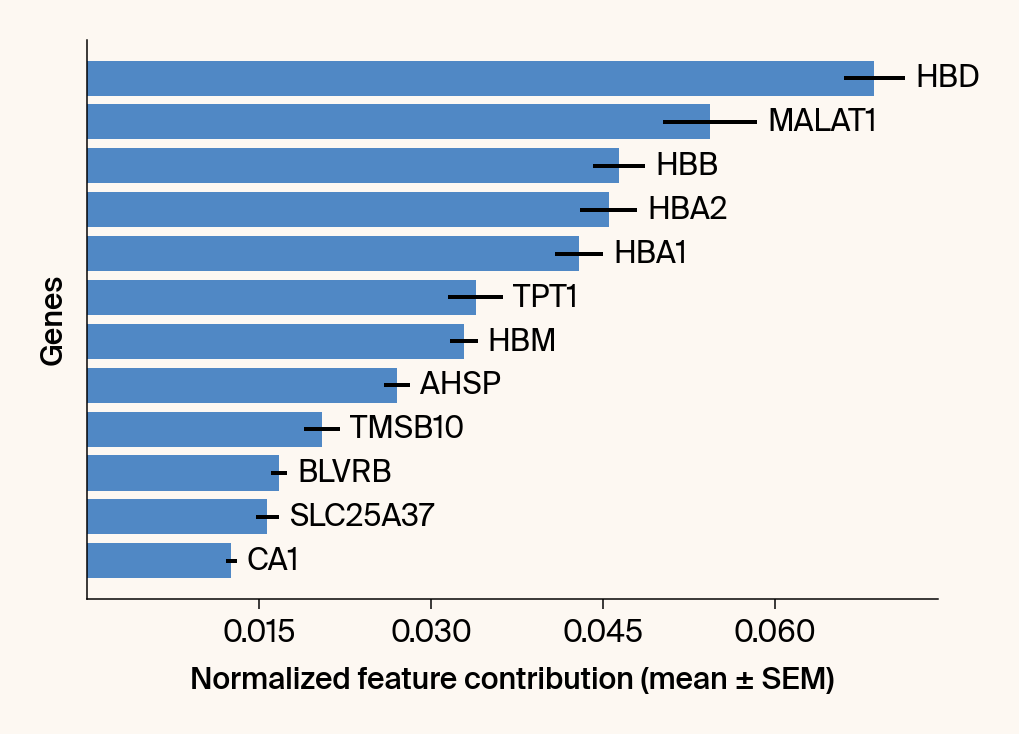
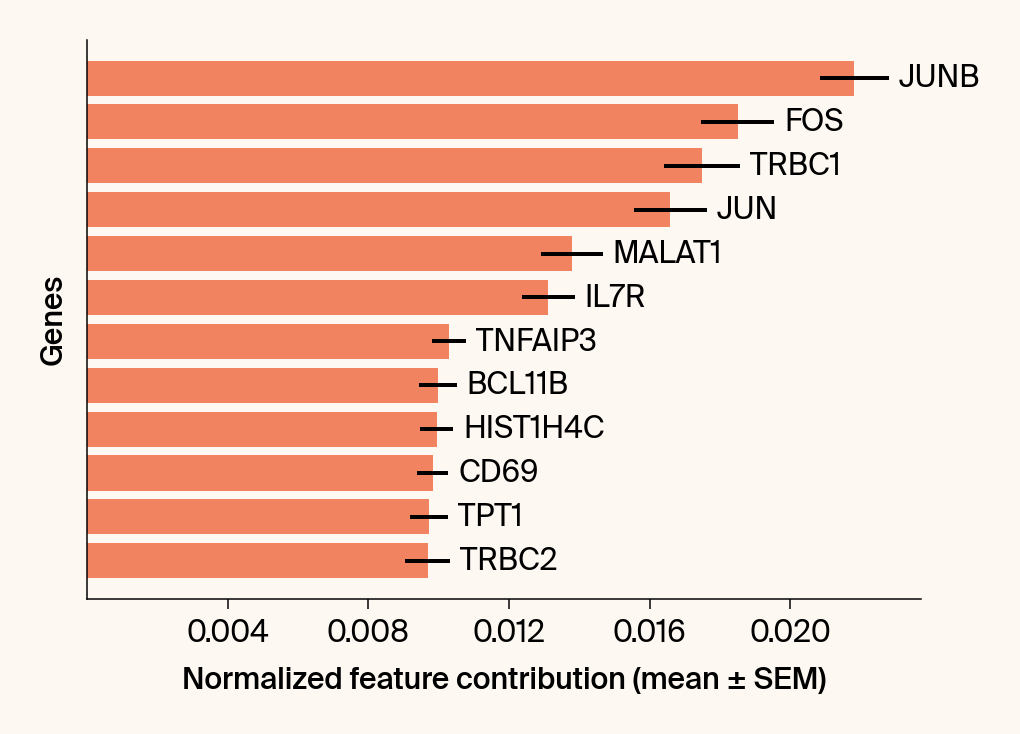
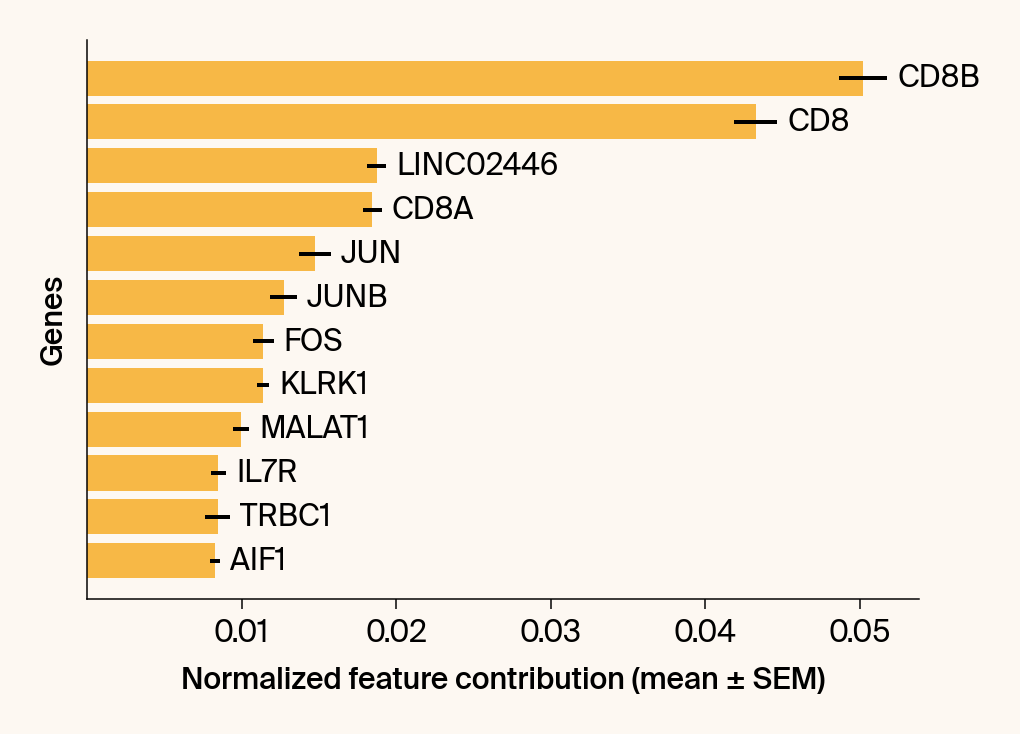
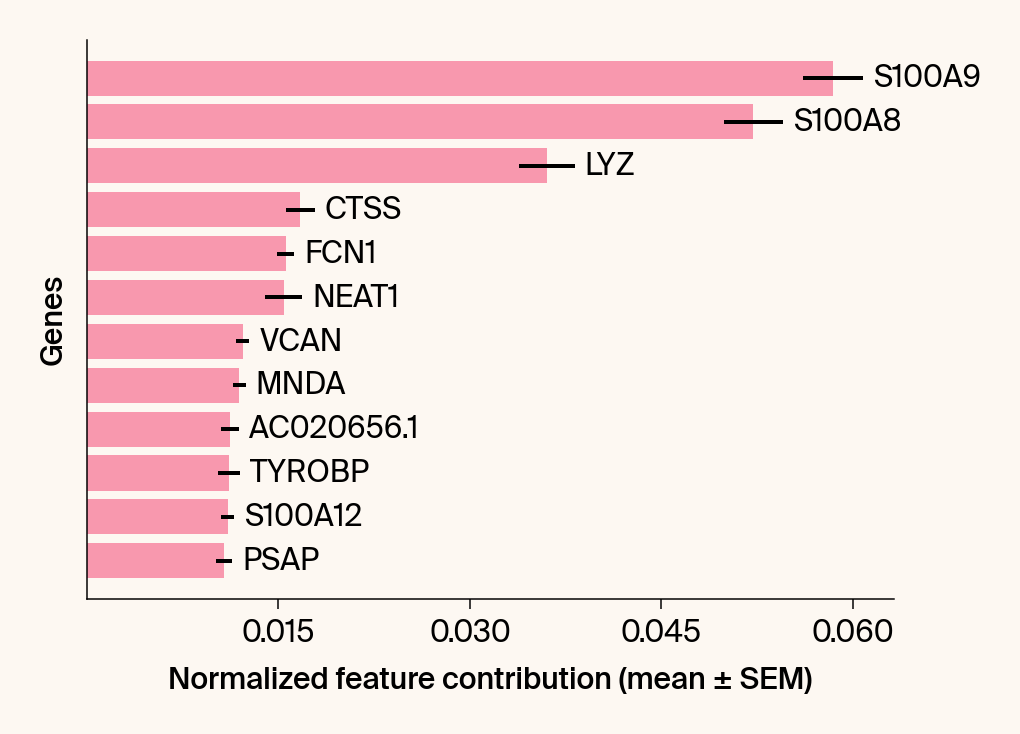
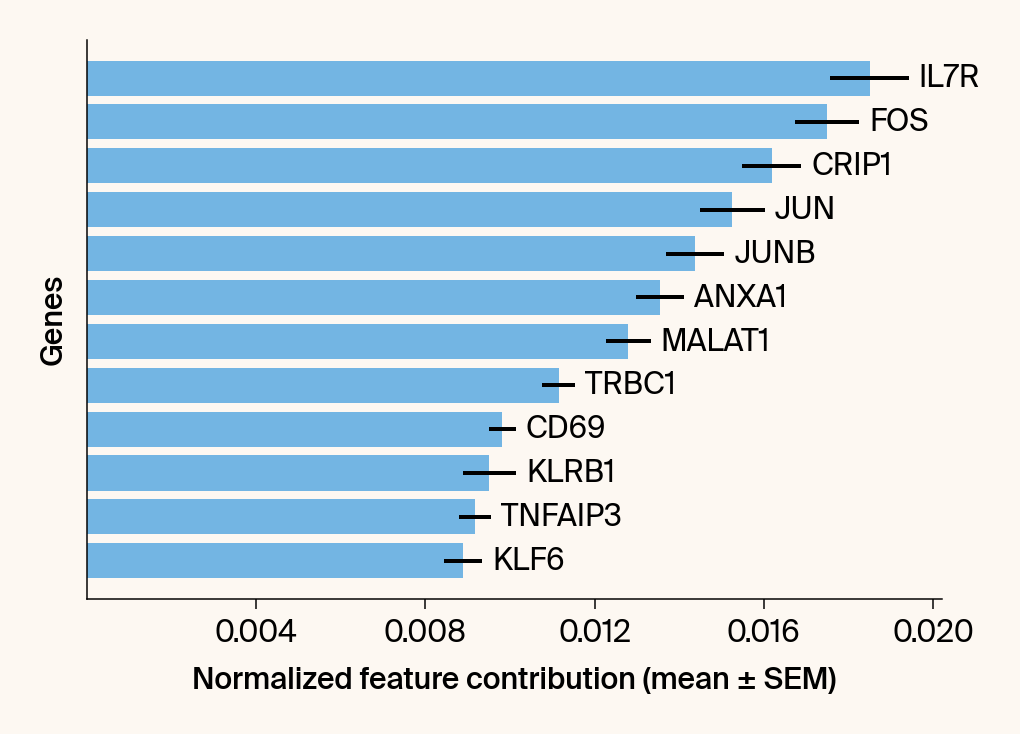
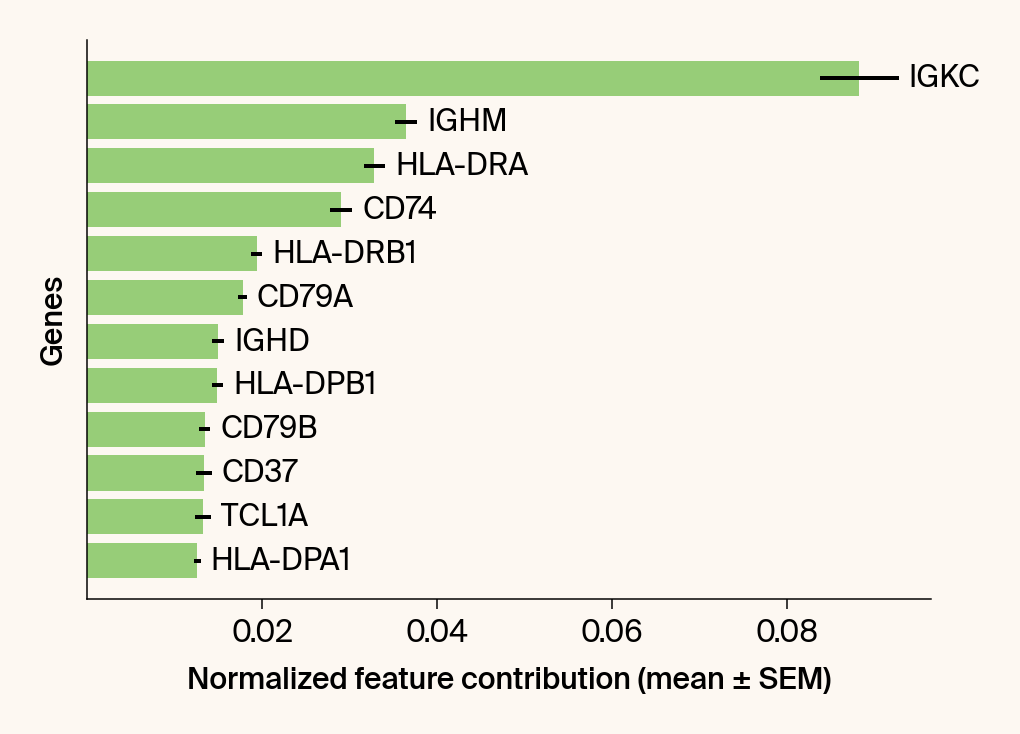
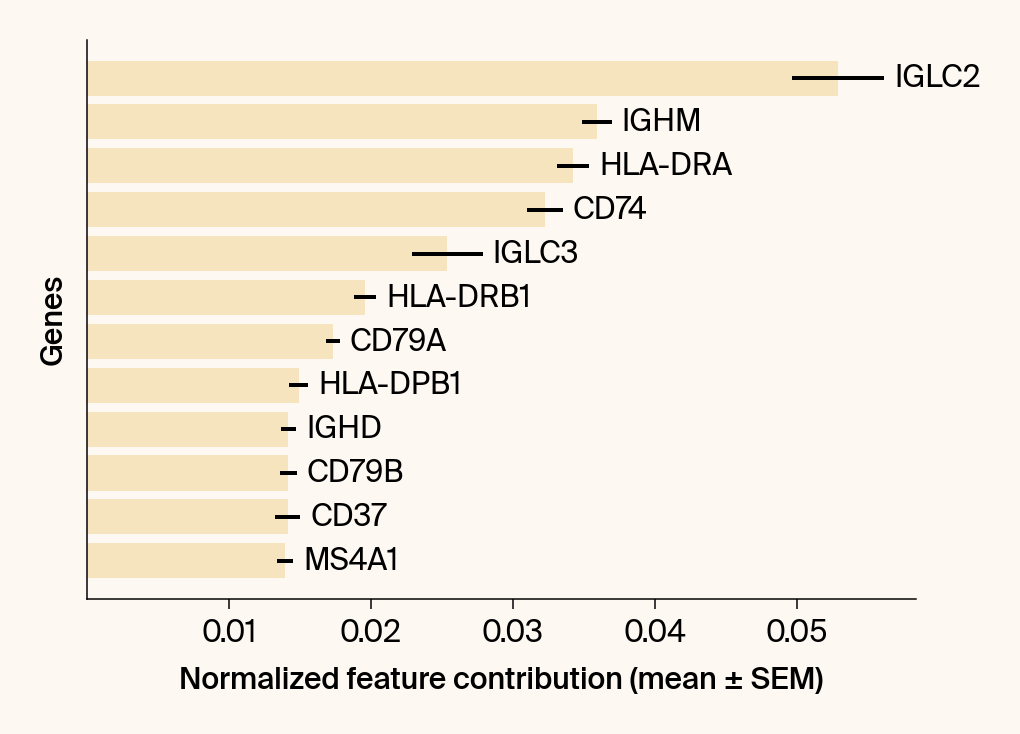
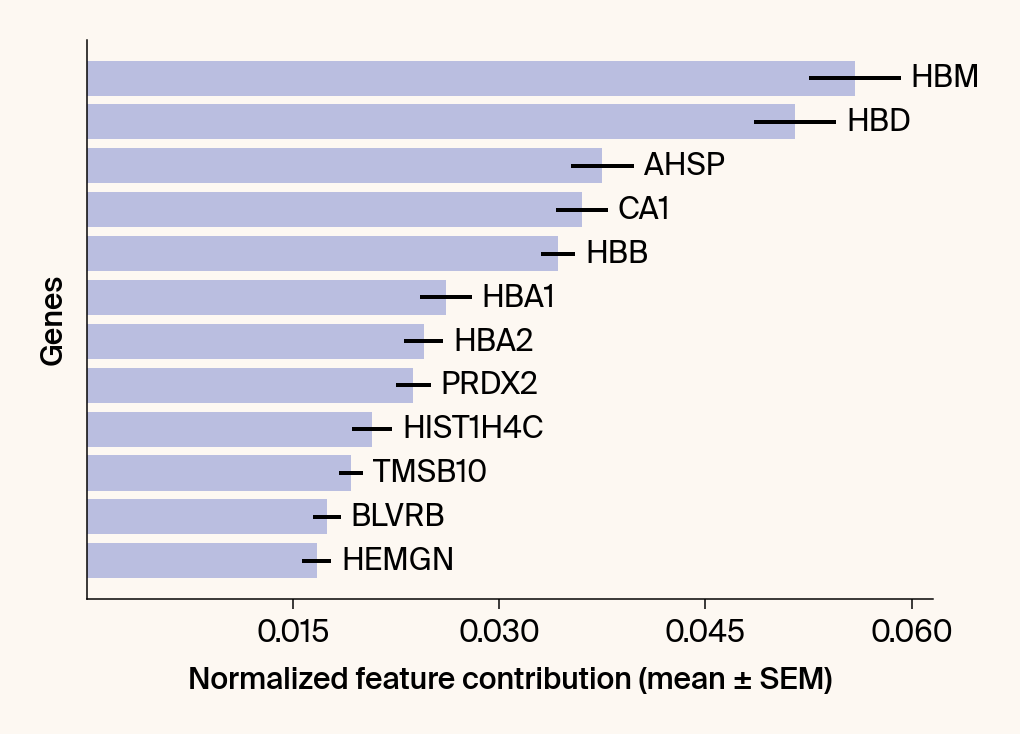
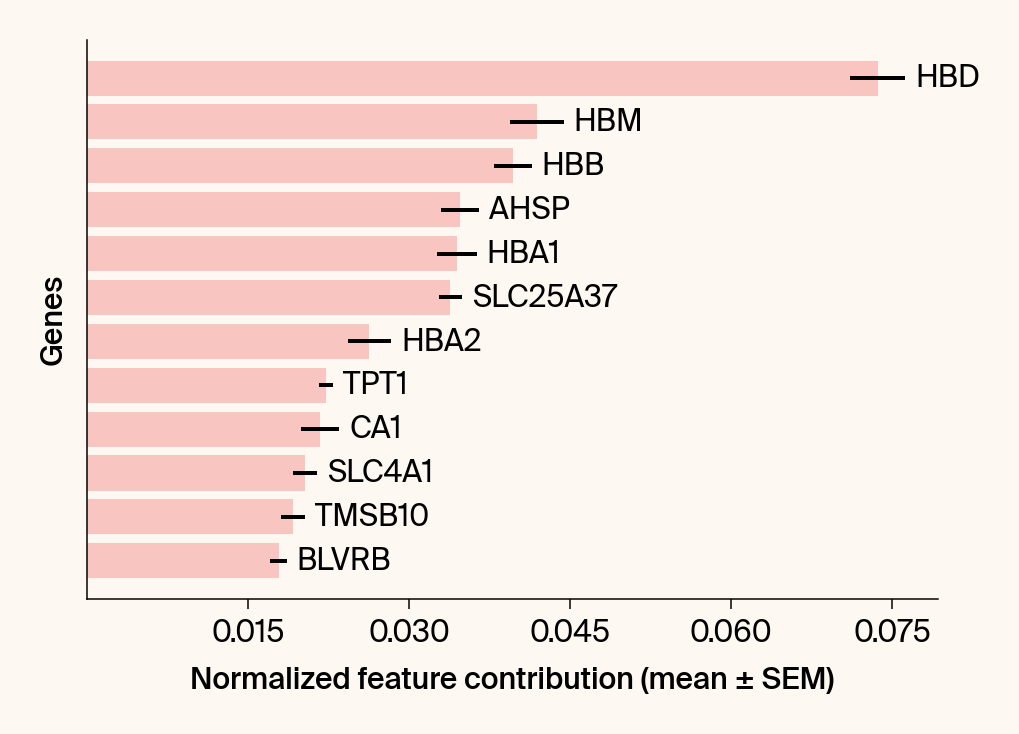
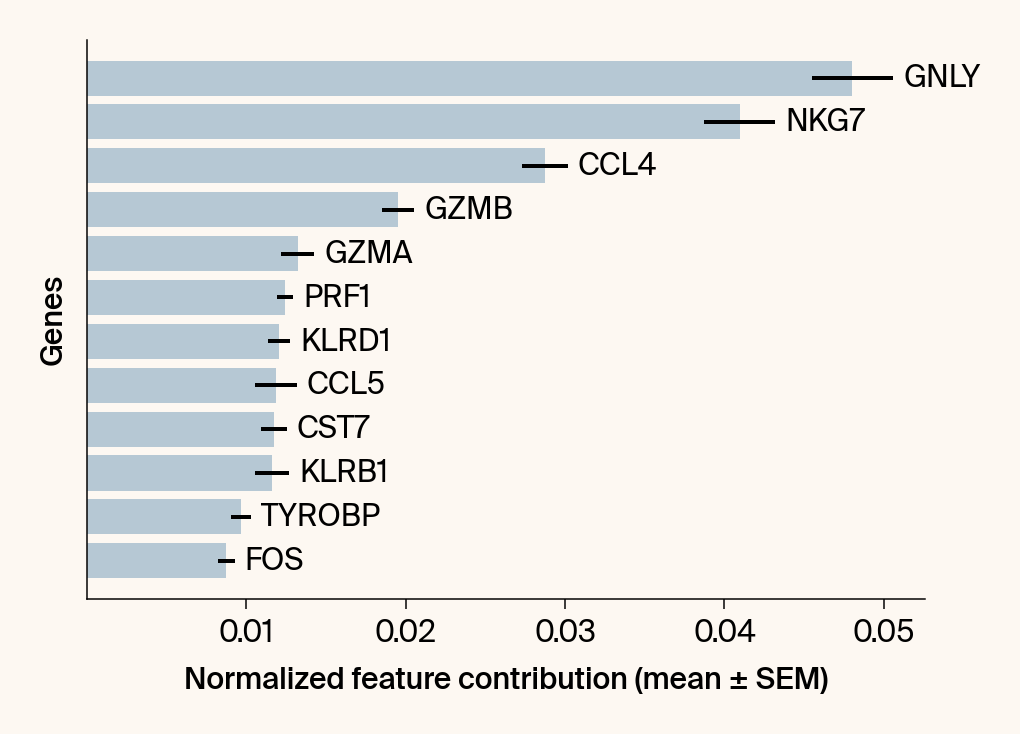
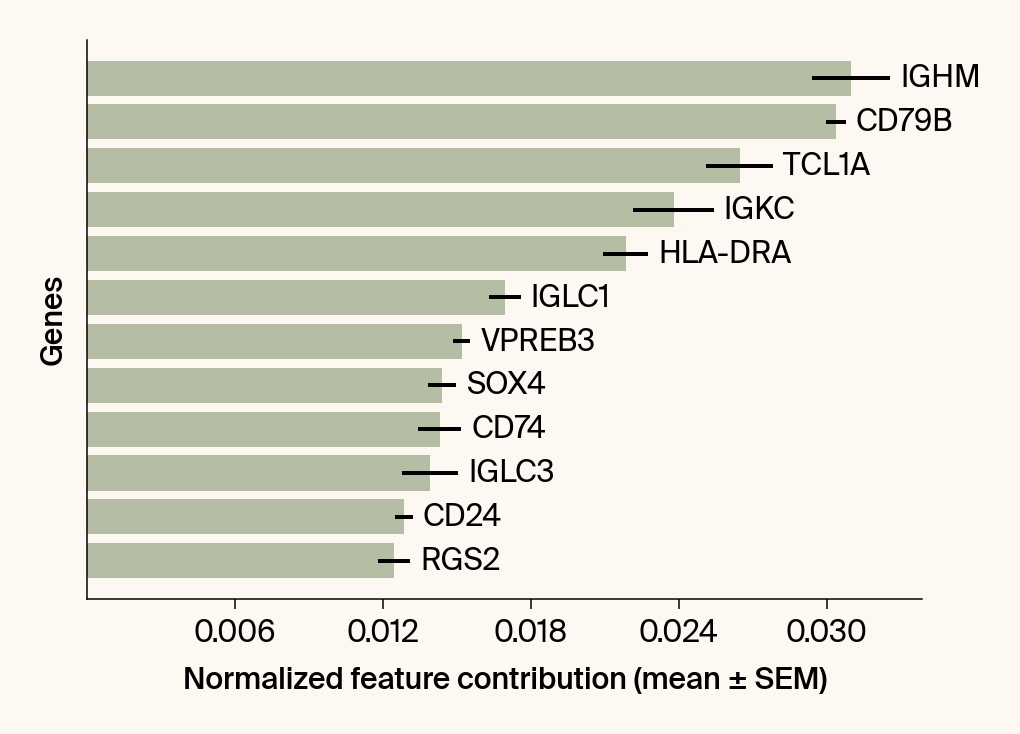
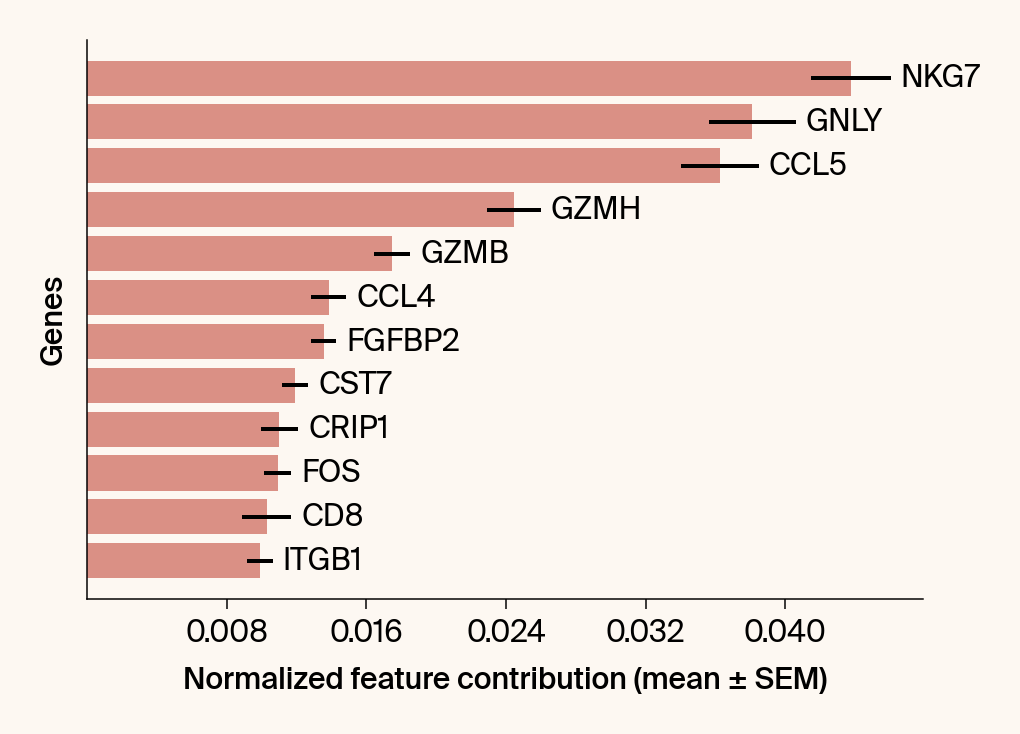
top_genes = group_df.loc[top_bar_indices, 'gene'][::-1]
top_means = group_df.loc[top_bar_indices, 'mean_contribution'][::-1]
top_sems = group_df.loc[top_bar_indices, 'sem_contribution'][::-1]
if plot_sum_features:
# Assuming n_features_vectors is at least 20, or you want the top 'n_features_vectors'
# Change the index to top 20 features, or use the existing variable
top_path_indices = group_df.index[:220]
# --- Initialize starting point and list for line segments ---
current_pos = np.array([0.0, 0.0])
path_points = [current_pos.copy()] # Start the path at (0, 0)
# --- Calculate the sequential path and plot vectors ---
for gene_idx in top_path_indices:
gene_name = group_df.loc[gene_idx, 'gene']
gene_original_index = gene_to_original_index.get(gene_name)
if gene_original_index is not None:
vec = vectors_for_group[:, gene_original_index]
# --- Plot the vector segment ---
# The vector starts at the end of the previous one (current_pos)
# and points to the new position (current_pos + vec)
plt.arrow(current_pos[0], current_pos[1], vec[0], vec[1],
width=0.15, color='r', head_width=0.0, zorder=4,
label='Sequential Path' if len(path_points) == 1 else None)
# --- Update position and path points ---
current_pos += vec
path_points.append(current_pos.copy())
# --- Add text label at the end of the vector segment ---
# plt.text(current_pos[0], current_pos[1], gene_name, fontsize=10, color='r',
# bbox=dict(boxstyle="round,pad=0.1", fc='white', ec="none", alpha=0.6))
# --- Plot the path as a single line for clarity (optional) ---
path_points_array = np.array(path_points)
plt.plot(path_points_array[:, 0], path_points_array[:, 1], 'r--', alpha=0.5, zorder=3)
fig1.tight_layout()
plt.show()
# --- Plot 2: Bar chart ---
# Create a new, separate figure
fig2 = plt.figure()#figsize=[6, 6])
# Use plt.barh
bars = plt.barh(top_genes, top_means, xerr=top_sems, capsize=3, color=color_map.get(group, 'gray'))
# Get current axis for bar_label, spines, and box_aspect
current_ax = plt.gca()
current_ax.bar_label(bars, labels=[f'{g}' for g in top_genes], padding=5)
# Use plt.tick_params, xlabel, ylabel
plt.tick_params(axis='y', left=False, labelleft=False)
plt.xlabel("Normalized feature contribution (mean ± SEM)")
plt.ylabel("Genes")
current_ax.spines[['right', 'top']].set_visible(False)
if set_axes_equal:
current_ax.set_box_aspect(1)
current_ax.xaxis.set_major_locator(plt.MaxNLocator(nbins=6, prune='both'))
fig2.tight_layout()
plt.show()
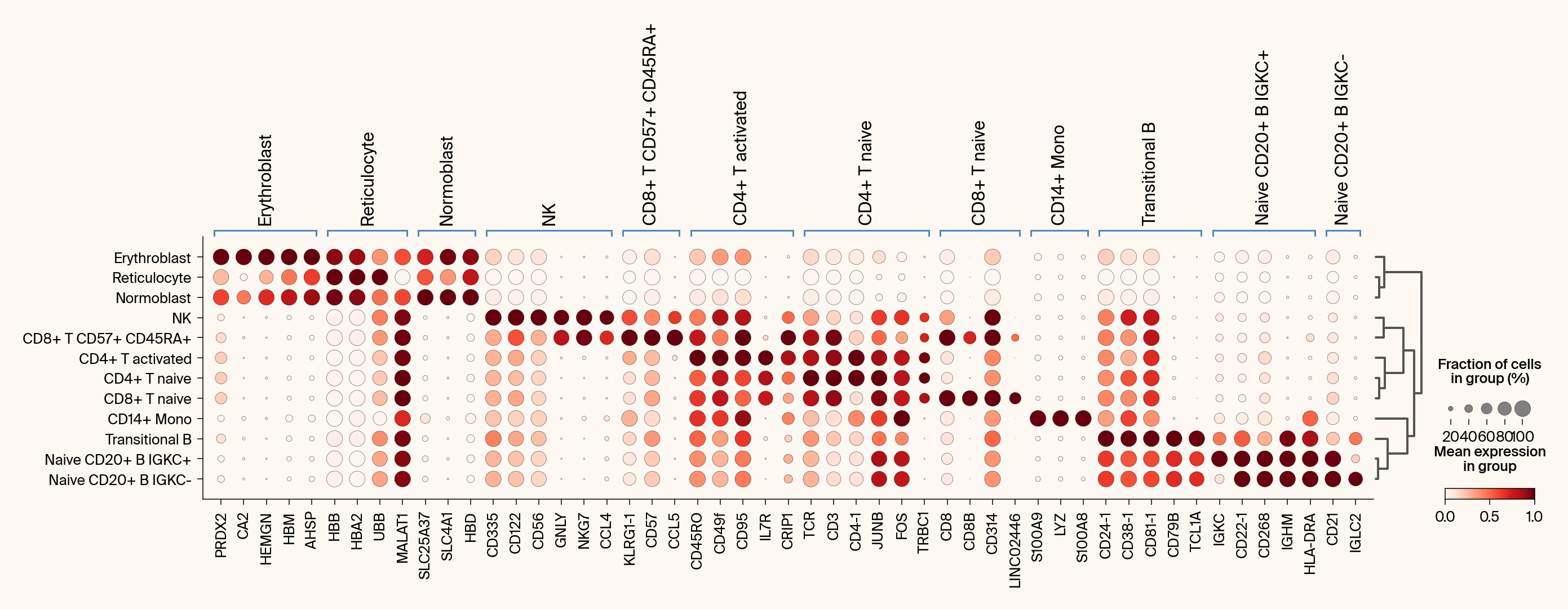
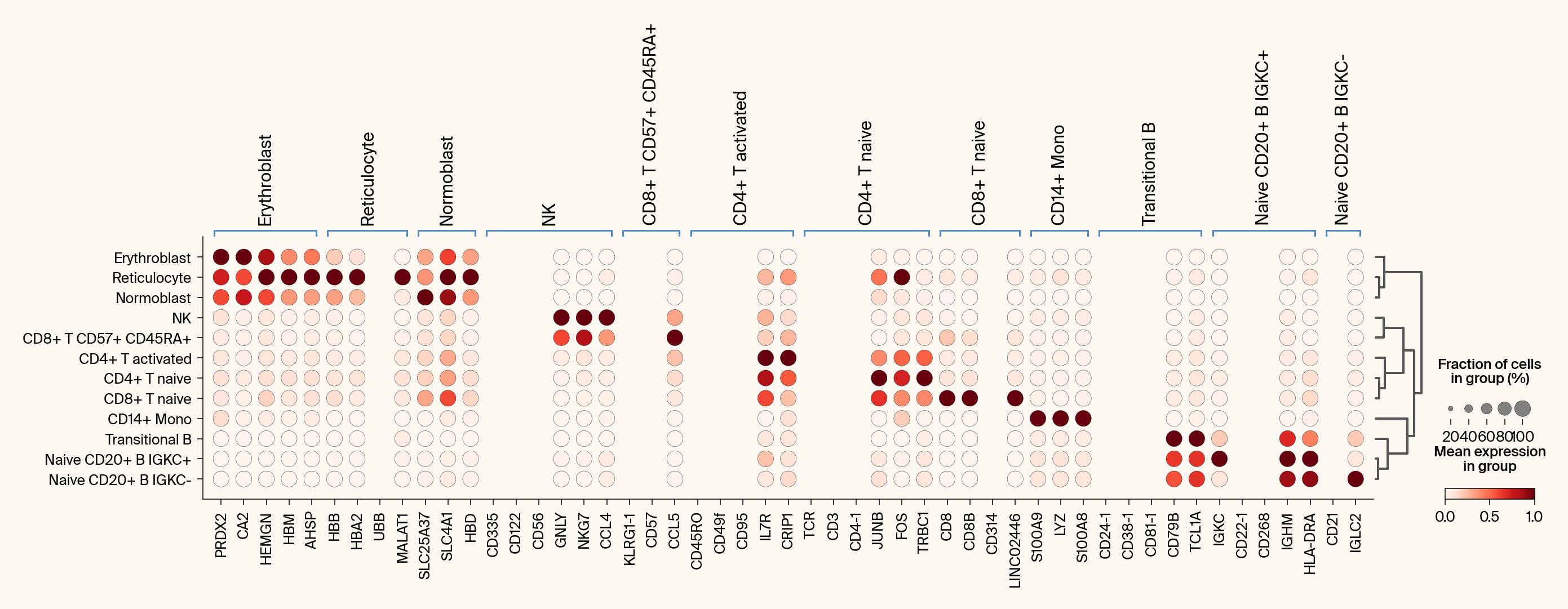
def compare_with_differential_expression(
reducer: GlassBoxUMAP,
adata: ad.AnnData,
groupby: str = 'cell_type',
n_top_genes: int = 2,
summary_stats_file: str = "analysis_summary_stats.csv",
summary_plot_file: str = "analysis_summary_plot_data.npz"
):
"""
Compares Jacobian features with differential expression via dot plots.
Args:
reducer (GlassBoxUMAP): The fitted model object.
adata (ad.AnnData): The processed AnnData object.
groupby (str): The .obs column to use for grouping (e.g., 'cell_type').
"""
import os
from collections import defaultdict
layer_to_use = None
can_load_from_file = (
summary_stats_file and os.path.exists(summary_stats_file) and
summary_plot_file and os.path.exists(summary_plot_file)
)
if can_load_from_file:
summary_df = pd.read_csv(summary_stats_file)
jacobian_dict = {}
for group in adata.obs[groupby].cat.categories:
if group in summary_df[groupby].values:
top_genes = summary_df[summary_df[groupby] == group].sort_values(
'mean_contribution', ascending=False
)['gene'].values[:n_top_genes]
jacobian_dict[group] = list(top_genes)
with np.load(summary_plot_file, allow_pickle=True) as data:
if 'jacobian_magnitude' not in data or 'gene_names' not in data:
print("Warning: 'jacobian_magnitude' or 'gene_names' not found in summary file.")
else:
loaded_magnitude = data['jacobian_magnitude']
loaded_genes = data['gene_names'] # Gene names from the NPZ file
# Re-align loaded layer with current adata
gene_to_npz_index = {gene: i for i, gene in enumerate(loaded_genes)}
new_layer = np.zeros(adata.shape, dtype=loaded_magnitude.dtype)
genes_found = 0
for i, gene in enumerate(adata.var_names):
if gene in gene_to_npz_index:
new_layer[:, i] = loaded_magnitude[:, gene_to_npz_index[gene]]
genes_found += 1
adata.layers['jacobian_magnitude'] = new_layer
layer_to_use = 'jacobian_magnitude'
else:
if not reducer.feature_contributions_:
raise RuntimeError("Must run compute_attributions() first to generate stats.")
stats_df = reducer.get_feature_importance(adata, groupby, adata.var_names.values)
jacobian_dict = {}
for group in adata.obs[groupby].cat.categories:
if group in stats_df[groupby].values:
top_genes = stats_df[stats_df[groupby] == group].sort_values(
'mean_contribution', ascending=False
)['gene'].values[:n_top_genes]
jacobian_dict[group] = list(top_genes)
jacobxall_first_run = reducer.feature_contributions_[0]
adata.layers['jacobian_magnitude'] = np.linalg.norm(jacobxall_first_run, axis=1)
layer_to_use = 'jacobian_magnitude'
sc.tl.rank_genes_groups(adata, groupby=groupby, method="wilcoxon", n_genes=n_top_genes)
de_dict = {
name: list(adata.uns['rank_genes_groups']['names'][name])
for name in adata.uns['rank_genes_groups']['names'].dtype.names
}
combined_genes = defaultdict(list)
seen_genes = set()
for d in (de_dict, jacobian_dict):
for key, value in d.items():
for gene in value:
if gene not in seen_genes:
combined_genes[key].append(gene)
seen_genes.add(gene)
with mpl.rc_context({"figure.facecolor": apc.parchment, "axes.facecolor": apc.parchment}):
# Plot 1: Differential Expression
ax1 = sc.pl.rank_genes_groups_dotplot(
adata, var_names=combined_genes, groupby=groupby,
standard_scale="var", show=False
)
# plt.title("Differential expression (Combined DE + Jacobian Genes)")
# plt.show()
# Plot 2: Jacobian Feature Importance
if layer_to_use:
ax2 = sc.pl.rank_genes_groups_dotplot(
adata, var_names=combined_genes, groupby=groupby,
layer=layer_to_use, standard_scale="var", show=False
)
# plt.title("Jacobian feature importance")
# plt.show()
else:
print("Warning: Could not plot Jacobian dot plot. 'jacobian_magnitude' data was not found.")
plt.show()
def set_fonts():
"""(Optional) Setup custom fonts."""
font_files = fm.findSystemFonts('Suisse Int_l/')
if len(font_files)>0:
for font_file in font_files:
fm.fontManager.addfont(font_file)
set_fonts()
setup_plotting_themes()